
We are the tech transfer platform that brings innovation into life science workflows. We produce the most innovative R&D tools, side by side with the researchers who design, test and use them.
We are the tech transfer platform that brings innovation into life science workflows. We produce the most innovative R&D tools, side by side with the researchers who design, test and use them.
)")
")
)")
")

")
")


")

")
SpheroTribe provides a complete toolkit for generating reproducible 3D cell structures (spheroids, organoids) without restrictions of cell type.
The kit includes:
– 5X methylcellulose solution
– U-bottom 96-well plates
– pipette tips with a large opening of 200µL
| Size/content | Catalog Number |
| 10x 96-well plates (flat bottom) 2x tip boxes 1 methylcellulose bottle | TDA-SPK-KIT |
| 1x 96-well plates (flat bottom) 20 tips 2,5 mL of methylcellulose | TDA-SPK-MINIKIT |
| 1 methylcellulose bottle | TDA-SPK-2-25 |
SpheroTribe provides a simple toolkit to generate consistent and robust 3D cell structures. Simply dilute the SpheroTribe solution into your culture medium of choice, watch your cells turn into uniformly sized 3D spheroids and collect them for your downstream assays.
Once diluted in your culture medium of choice, our concentrated polymer-based solution increases the medium viscosity favouring cell-cell contacts. SpheroTribe offers a simple method to generate homogeneous 3D cell structures with increased control over their size and shape, which can be easily handled and washed for downstream experiments.
SpheroTribe is particularly useful to boost aggregation when working with challenging cells, minimize variability between samples and improve the consistency of your migration/invasion assays, immunostaining, drug screening or in vivo implantation experiments.
In addition to the SpheroTribe solution, the full kit also includes U-bottom plates and wide-opening tips so you have everything you need to get started with your 3D cell culture experiments.
25mL kit contents:
– 25mL of 5X methylcellulose solution
– 10x U-bottom 96-well plates
– 2x racks of 96 pipette tips (200µL) with a large opening
2,5mL kit contents:
– 2.5mL of 5X methylcellulose solution
– 1x U-bottom 96-well plate
– 20 pipette tips (200µL) with a large opening
Recommendation for the methylcellulose solution:
4°C for (at least) 6 months.

SpheroTribe provides a gel-like scaffold that favors cell-cell contacts by increasing medium viscosity. It was shown to generate compact spheroids mimicking solid tumors, improve spheroid formation with some of the most challenging cells and to speed up stem cell-derived organoid formation.
By maximizing cell aggregation, SpheroTribe promotes the formation of unique & uniformly sized spheroids allowing for consistent assays (growth, invasion, immune infiltration, in vivo injection, etc).
No need to work on ice, have access to sophisticated equipment of expertise. With SpheroTribe, you have everything on hand to easily grow & handle your spheroids.
The SpheroTribe solution is composed of methylcellulose, a biologically inert compound dissolved in basal culture medium without any proteins, lipids or growth factors. You can dilute it in any culture medium of your choice, and add additional compounds as desired (i.e. serum, antibiotics, differentiation factors, etc).
Patient-derived stem-like glioblastoma cells (GB P3 and BL13), human glioblastoma cell lines (U87 & T98G), HeLa, human vaginal mucosal melanoma (HMV-II), human primary colorectal cancer cells, human breast cancer cells (MDA-MB 231), human induced pluripotent stem cells, monkey kidney fibroblast-like cell line (COS-7), primary neurons from rat embryos (E18) & murine melanoma cells (B16F10).
Once spheroids have grown to your desired size, you can use them for any kind of assay according to your regular workflow. The SpheroTribe solution can be readily washed off, leaving a spheroid available for other tests at any stage of your protocol.
Example of in situ assays you can perform directly on the U-bottom plate supplied:
Examples of downstream assays that might require transferring spheroids to other vessels:
For more guidance on downstream assays, check out our FAQ section containing useful tips & example protocols.


Human glioblastoma U87 cells were cultured in DMEM with or without SpheroTribe
in U-bottom plates and imaged after 2 days (4X magnification) and 6 days (10X magnification)

Glioblastoma spheroids were included into a collagen matrix after being cultured in medium added with SpheroTribe solution for 4 days.
Images were taken immediately after (left) or 24 hours after (right) inclusion in the collagen matrix.
Image credits: (c) Thomas Daubon, 2023.

A. 10,000 B16F10 cells were grown for 6 days as spheroids using SpheroTribe. B. 100,000 PBMC from murine spleen were activated with IL-15 (40 ng/mL) [1], incubated with anti-PD1 (10 µg/mL) for 1h and added on B16F10 spheroids for 3 days.
Graph shows flow cytometry quantification of differential lymphocyte infiltration after spheroid dissociation according to treatment. N=4. Mann-Whitney U Test, p-value<0.05. [1] https://doi.org/10.3389/fonc.2022.898732.
Image credits: Guillaume Mestrallet, PhD – Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, US – 2023. @GMestralletPhD

Human iPSCs were seeded in ULA plates in presence (left) or absence (right) of SpheroTribe solution (T=0) and cultured for 3 days following a self-assembling human heart organoid differentiation protocol [1].
Image credits: Aitor Aguirre, Ph. D. – Michigan State University, US
[1] Lewis-Israeli, Y.R., Wasserman, A.H., Gabalski, M.A. et al. Self-assembling human heart organoids for the modeling of cardiac development and congenital heart disease. Nat Commun 12, 5142 (2021). https://doi.org/10.1038/s41467-021-25329-5

U-87 glioblastoma cells were seeded at 1,000 cells per well in round-bottom wells molded in agarose using a Stampwell U-shape (Idylle) in full culture medium without (right) or added with SpheroTribe (left). After 3 days, pictures were taken and number of cell aggregates per well were quantified from 64 independent wells and associated standard deviation (SD) values were calculated.

COS-7 cells were cultured in non-tissue culture treated U-bottom plates for 4 days in presence (up)
or absence (bottom) of the SpheroTribe concentrate. Scale bar = 500 µm

HeLa cells were cultured in non-tissue culture treated U-bottom plates for 4 days in presence (up)
or absence (bottom) of the SpheroTribe concentrate. Scale bar = 500 µm

Need advice for your downstream assays? Check out our FAQ section for published protocols of immunostaining, invasion, migration, proliferation assays & in vivo spheroid implantation.
SpheroTribe relies on the use of methylcellulose, a crowding agent that is well-known to drive efficient cell aggregation into spheroids. Once diluted in culture medium, methylcellulose increases the viscosity of the medium, creating a gel-like environment that restrict cell movement and encourages cell-cell contacts for the formation of compact spheroids.
The SpheroTribe solution is composed of methylcellulose, a derivative of cellulose that is known to be biologically-inert, i.e. to neither actively contribute to biological responses or interfere with it. It is notably used in various biofabrication tools, including bioinks. In addition, SpheroTribe allows you to culture cells without exogenous ECM components, avoiding the known bias they may have on cell signaling. If needed, SpheroTribe can be readily washed off at any stage of the protocol to leave spheroids available for subsequent assays.
So far, SpheroTribe has been successfully used to generate spheroids/organoids with the following cell types:
Patient-derived stem-like glioblastoma cells (GB P3 and BL13), human glioblastoma cell lines (U87 & T98G), HeLa, human vaginal mucosal melanoma (HMV-II), human primary colorectal cancer cells, human breast cancer cells (MDA-MB 231), monkey kidney fibroblast-like cell line (COS-7), primary neurons from rat embryos (E18) & murine melanoma cells (B16F10), human induced pluripotent stem cells (hiPSCs)-derived organoids.
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
The SpheroTribe concentrated solution is made up in basal medium and does not contain any proteins, lipids or growth factors. You can dilute it in any culture medium of your choice, and add other compounds as desired (i.e. serum, antibiotics, etc).
One SpheroTribe kit contains everything you need to grow up to 960 unique spheroids. Total number of spheroids generated with one kit may vary depending on the concentration of SpheroTribe solution used for cell aggregation, total spheroid growth duration and frequency of medium renewal.
Yes, it is also possible to buy the methylcellulose solution alone and use it to complement your current method. Please contact us and we will send a dedicated quote.
The total time to initiate 3D cell culture with SpheroTribe should take no more than 30 minutes. After that, optimal culture duration to achieve desired spheroid size will vary depending on your cell proliferation rate and targeted application. As a general rule, compact & homogeneous spheroids are usually formed after 3 days of culture.
Optimal seeding density can vary greatly depending on the cell type, proliferation rate and targeted application. You will most probably need to start with a couple of optimization tests to find out which density will best suit your needs. As a general rule, we recommend using 10,000 to 20,000 cells per well for primary cells and 2,000 to 7,000 cells per well for tumoral/immortalized cell lines.
Spheroid sizes can vary greatly depending on cell type characteristics, initial density and culture conditions used. To help you select optimal conditions, here are a couple of examples of sizes reached for different cell types and seeding densities:
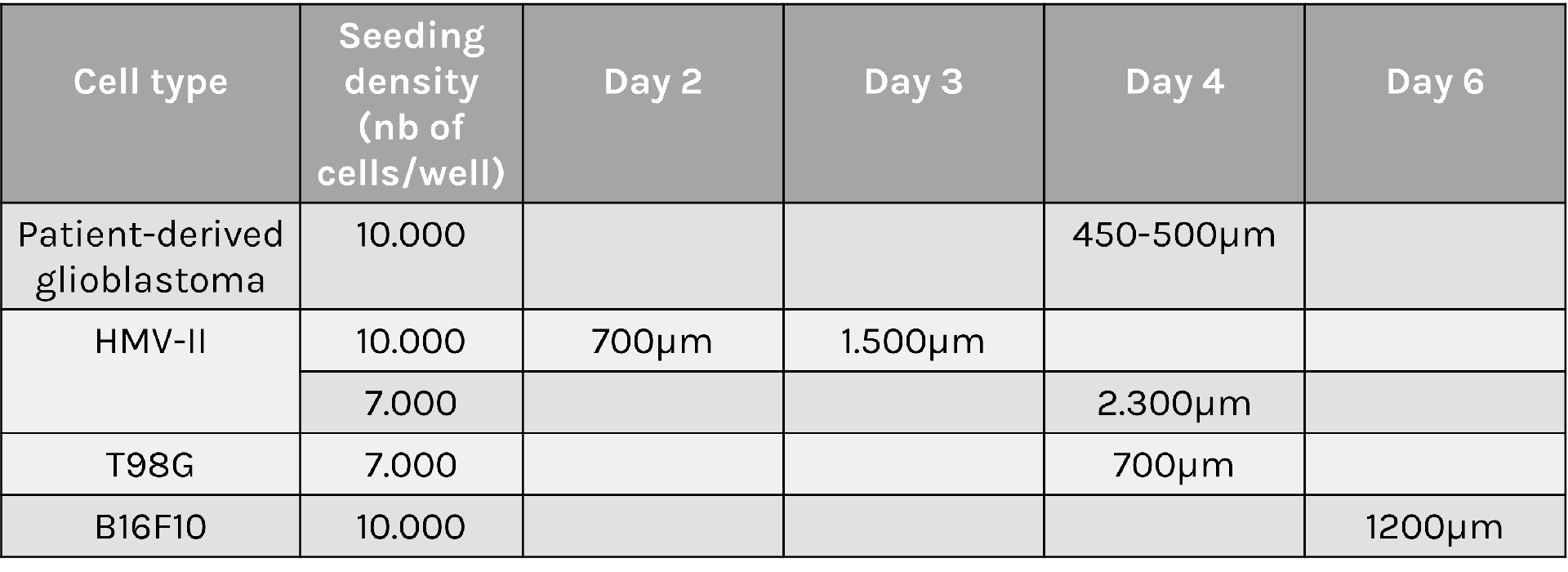
A variety of downstream assays have been performed with spheroids generated with SpheroTribe. We only provide a detailed protocol for the formation of spheroids, as this is what SpheroTribe is designed for. However, if needed, you can check out the following publications describing protocols for various assays using glioblastoma spheroids generated with SpheroTribe. These can easily be applied to other cell types:
The SpheroTribe kit has been shown to generate unique & uniform spheroids with most cell types tested. Yet, when working with particularly challenging cells, here are a few tips to keep in mind to maximize your chance to obtain unique & uniform spheroids in each well:
Any other questions? Please contact us
Original publication:
Other publications using SpheroTribe spheroids:
Chitosan-coated coverslips for live bacteria imaging
Chitozen is a chitosan-coated coverslip immobilizing bacteria for microscopy without inducing bacteriostatic effects. It is compatible with 6-channel sticky slides for flow experiments. It is very helpful if you want to:
Assay compatibility:
> Fluorescence
> Co-cultures (bacterial predators, immune cells)
> Addition of external factors (e.g. antibiotics, chemicals, inhibitors)
> Static or dynamic conditions
Imaging modes:
> phase-contrast,
> epifluorescence,
> confocal,
> super-resolution microscopy,
> atomic force microscopy (AFM) – documentation & example pictures available on request
Experimental outputs:
> Behavioural changes: growth, elongation, cell division, fitness, colony/biofilm formation, etc
> Single molecule imaging
Chitozen is efficient with the following bacteria:
> E. coli
> Bacillus subtilis
> Caulobacter crescentus
> Corynebacterium glutamicum
> Helicobacter pylori
> Mycobacterium smegmatis
> Myxococcus xanthus
> Pseudomonas aeruginosa
> Pseudomonas fluorescens
> Salmonella
> Staphylococcus aureus
> Vibrio cholerae
This list is updated regularly according to feedback provided by researchers who use Chitozen.
Chitozen coverslips are compatible with 6-channel sticky slides to work in a closed system in static or dynamic flow conditions. Order you coverslips alone if you need to work in an open system (i.e. for AFM imaging).
5-coverslips only*
> 5x standard (25 x 75 mm) chitosan-coated coverslips
5-coverslips with microfluidic channels
> 5x standard (25 x 75 mm) chitosan-coated coverslips
> 5x bottomless 6-channel sticky slides
(Optional) centrifugation pack: the Chitozen coverslips can be centrifuged using a rack compatible with standard microscope slide (25mmx75mm) dimensions. If needed, add our centrifugation pack to your Chitozen kit. It includes:
> µ-Slide Microscopy Rack (ibidi
> Magnetic Lid for Microscopy Rack
> Clamp & adapter for sticky slides
*If using the Chitozen coverslips alone, separate wells can be created manually by using a sealing glue or Stencell silicon chambers. We can provide these 2 products for you. Contact us for more information.
For custom orders (i.e. alternative coverslip amounts), please contact us.
Lifetime: up to 12 months at room temperature, shielded from direct sunlight
© 2019 Tréguier et al. This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.

Credit: Amandine Desorme, LCB – CNRS, 2021
Credit: Amandine Desorme, LCB – CNRS, 2021

Peptidoglycan is labeled with the green fluorophore BADA.
Credit: Amandine Desorme, LCB – CNRS, 2021

AB1157 E. coli expressing DNA marker HU-mCherry were imaged at 37oC with perfusion of ½ LB at a flow rate of 2 ml/min. Drug X was added at time point 35 minutes and removed at time point 60 minutes.
Credit: Emily Helgesen – Oslo University Hospital – 2022

AB1157 E. coli expressing SeqA-YFP (green), a protein associated with DNA replication, were imaged at 37°C over 60 minutes without perfusion of medium
Credit: Emily Helgesen – Oslo University Hospital – 2022
E. coli BW25113 cells were imaged at 37°C with perfusion of M9 medium at a flow rate of 0.05 mL/min.
Credit: Bianca Sclavi – 2021

E. coli MG1655 with chromosomally encoded HU-eGFP under the native promoter were imaged with perfusion at a flow rate of 0.05 mL/min.
Credit: Itzhak Fishov – Ben-Gurion University of the Negev, 2022
A technology designed by Tâm Mignot, Olivier Theodoly, Amandine Desorme, Guillaume Sudre and Laurent David.
Everspark buffer at 100 mM MEA in Tris buffer
10 vials containing 450 μL each (total volume 4,5 mL)

| Description | Catalog Number |
| Everspark 1.0 | KMO-ETE-450 |
| Everspark 2.0 | KMO|ETE+|450-10 |
Discover the long-lasting Everspark blinking buffer series for prolonged blinking in dSTORM microscopy. No more rushing your imaging. Mount your sample, and get stable blinking for weeks.
NEW ! Discover the newly-developed Everspark 2.0 also compatible with green fluorophores (i.e. AF488) for improved resolution in 3-colour dSTORM microscopy.
Compatible fluorophores: yellow to far-red
Validated fluorophores include JF 549, DL 550, CF 555, CF 568, AF 568, JF 646, AF 647, CF 647, Atto 647N, DL 650, CF 680 & Cy5
Multicolor imaging: +
Blinking stability (after mounting): up to 2 months
Blinking events: +
Compatible fluorophores: green, yellow & far-red
Validated fluorophores include AF 488, MemBright 488, FITC, FAM, Spy555, CF 568 & AF 647
Multicolor imaging : +++
Blinking stability (after mounting): up to 3.5 months
Blinking events: +++
Kit description:
10 vials with 450 µL of Everspark buffer at 100 mM MEA in Tris.
1 vial per experiment.
Vials lifetime: Up to 6 months in a closed pouch (eg unmounted).
Laser requirements: for green fluorophore imaging (Everspark 2.0 only), we recommend using a 488 laser >200mW, ideally 500mW for optimal results.

Centrosomal proteins Cep152, rootletin and PCM labelled with AF647, CF568 & AF488 respectively were imaged on a Vutara VXL (Bruker) inEverspark 2.0 buffer before 3D dSTORM reconstruction
Credits: Karine Monier, INMG-PGNM, Lyon

Centrosomal proteins Cep152, rootletin and PCM labelled with AF647, CF568 & AF488 respectively were imaged on a Vutara VXL (Bruker) in Everspark 2.0 buffer before 3D dSTORM reconstruction
Credits: Karine Monier, INMG-PGNM, Lyon

AF488-coated beads were mounted in Everspark 1.0 or Everspark 2.0 buffer and imaged on a Vutara VXL (Bruker) before 3D dSTORM reconstruction.
Credits: Karine Monier, INMG-PGNM, Lyon

Left: Two centrosomes imaged the same Day (D0) and 7 days after mounting (D7) on the same slide stored in the dark at 4°C. The typical 450 nm donut-like structure is visualised using a colour-coded scale encoded with the IGOR software, where each point appears as a function of its localisation precision (5 to 60 nm; inverted rainbow colour scale). Labeling: Distal-appendages detected by immunofluorescence with AF647 in RPE-1 cells.
Right: The number of blinking events per centrosome and the median of the localization precision in nm are presented for each serie of 50,000 images recorded at D0 and D7 (left).
Credits: Camille Fourneaux & Karine Monier, CRCL, Lyon

Each point is represented by its centroid (purple points) and its gaussian width (white). Intensity profil is displayed on the right with the measurement from peak to peak, in agreement with the size of the distal appendage crone. Labeling: distal-appendages detected by immunofluorescence with AF647 in RPE-1 cells.
Credits: Corentin Rousset, Karine Monier, CRCL Lyon
Find below the protocol of use of our Everspark buffers:
Video Protocols:
Everspark technology has been intially developed by Karine Monier, Arnaud Favier and Christophe Place and published in Scientific Reports:
Provost, A., Rousset, C., Bourdon, L. et al. Innovative particle standards and long-lived imaging for 2D and 3D dSTORM. Sci Rep 9, 17967 (2019). https://doi.org/10.1038/s41598-019-53528-0
Publications:
Dia2 formin controls receptor activity by organizing plasma membrane lipid partitioning at the nanoscale
Changting LI, Yannick HAMON, David MAZAUD, Pamela GONZALEZ TRONCOSO, Marie DESSARD, Hai-tao HE, Christophe LAMAZE, and Cedric M. BLOUIN. bioRxiv 2023.12.15.571857; doi: https://doi.org/10.1101/2023.12.15.571857
Nanoscale engagement and clusterization of Programmed death ligand 1 (PD-L1) in the membrane lipid rafts of Non-Small Cell Lung Cancer cells
Martina Ruglioni, Simone Civita, Tiziano Salvadori, Sofia Cristiani, Vittoria Carnicelli, Serena Barachini, Iacopo Petrini, Irene Nepita, Marco Castello, Alberto Diaspro, Paolo Bianchini, Barbara Storti, Ranieri Bizzarri, Stefano Fogli and Romano Danesi bioRxiv 2022.08.09.503318; doi: https://doi.org/10.1101/2022.08.09.503318
HIV-1 diverts actin debranching mechanisms for particle assembly and release in CD4 T lymphocytes
Rayane Dibsy, Erwan Bremaud, Johnson Mak, Cyril Favard, Delphine Muriaux
Nat Commun. 2023 Oct 31;14(1):6945. doi: 10.1038/s41467-023-41940-0
Fluorescent Polymer-AS1411-Aptamer Probe for dSTORM Super-Resolution Imaging of Endogenous Nucleolin
Fabre L, Rousset C, Monier K, Da Cruz-Boisson F, Bouvet P, Charreyre MT, Delair T, Fleury E, Favier A. Biomacromolecules. 2022 May 12. doi: 10.1021/acs.biomac.1c01706. PMID: 35549176
Comparative analysis of ChAdOx1 nCoV-19 and Ad26.COV2.S SARS-CoV-2 vector vaccines.
Michalik S, Siegerist F, Palankar R, Franzke K, Schindler M, Reder A, Seifert U, Cammann C, Wesche J, Steil L, Hentschker C, Gesell-Salazar M, Reisinger E, Beer M, Endlich N, Greinacher A, Völker U. Haematologica. 2022 Apr 1;107(4):947-957. doi: 10.3324/haematol.2021.280154. PMID: 35045692
Metabolic biorthogonal labeling and dSTORM imaging of peptidoglycan synthesis in Streptococcus pneumoniae
Jennyfer Trouve, Oleksandr Glushonkov and Cecile Morlot
Star Protocols, December 13, 2021. doi: 10.1016/j.xpro.2021.101006
Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia.
Greinacher A, Selleng K, Palankar R, Wesche J, Handtke S, Wolff M, Aurich K, Lalk M, Methling K, Völker U, Hentschker C, Michalik S, Steil L, Reder A, Schönborn L, Beer M, Franzke K, Büttner A, Fehse B, Stavrou EX, Rangaswamy C, Mailer RK, Englert H, Frye M, Thiele T, Kochanek S, Krutzke L, Siegerist F, Endlich N, Warkentin TE, Renné T.
Blood. 2021 Dec 2;138(22):2256-2268. doi: 10.1182/blood.2021013231. PMID: 34587242
Superresolution Microscopy of Drosophila Indirect Flight Muscle Sarcomeres.
Szikora S, Novák T, Gajdos T, Erdélyi M, Mihály J. Bio Protoc. 2020 Jun 20;10(12):e3654. doi: 10.21769/BioProtoc.3654. eCollection 2020 Jun 20. PMID: 33659324
Calibration beads for super-resolution microscopy 1 vial of 50 µL of a SpheroRuler suspension in PBS pH 7.4
| Description | Catalog Number |
| 10x 96-well plates (flat bottom) 2x tip boxes 1 methylcellulose bottle | TDA-SPK-KIT |
| 1x 96-well plates (flat bottom) 20 tips 2,5 mL of methylcellulose | TDA-SPK-MINIKIT |
| 1 methylcellulose bottle | TDA-SPK-2-25 |
SpheroRuler is a monodispersed suspension of 1µm diameter spheres coated with 647-fluorophores giving a stable blinking in SMLM microscopy. Their consistent size and geometry make them very practical and reliable standards to assess the accuracy of your x-y or z measurements, 3D reconstruction methods or your image quality. Monodispersed and immobile in solution, they can also be used as rulers, for drift correction or as guides to help localize features on your biological samples.
")
Kit description: 1 vial of 50 µL of a SpheroRuler suspension in PBS pH 7.4.
Allows 10 experiments when using the recommended 5µL volume in 35mm glass-bottom dishes.
Concentration: 7.10exp8 particles per mL.
Stability: up to 7 months when stored at 4°C.
Dye: 647-fluorophore (far-red fluorescence).
Compatible with: dSTORM, SRRF, Airyscan confocal, confocal, SEM
Following the success of SpheroRuler beads, we are thrilled to introduce some brand-new SpheroRulers of various sizes and colours that are now ready to join our catalog:

Interested in one or several of them? Please contact us


(A, B). Pictures of SpheroRuler beads acquired in wide-field (A) and SRRF-Stream super-resolution (B) imaging. Scale bar = 5µm
(C, D) Local enlargements of A and B pictures respectively.
Scale bar = 1µm
(E) Fluorescence intensity distributions along the solid lines in C and D.
Credits: Yao Baoli – Xi’an Institute of Optics and Precision Mechanics, Chinese Academy of Sciences, 20lksehg

SpheroRuler beads were imaged on a confocal microscope with a 63X oil objective.
Credits: Kseniya Korobchevskaya – Institute of Developmental & Regenerative Medicine, University of Oxford, UK
Credits: Lydia Y Li, Nabi Lab, The University of British Columbia, 2023
The SpheroRuler beads are 1μm-diameter polymer particles surrounded by 647-fluorophores covalently anchored to their surface. Beads will be visible as hollow rings or spheres when reconstructed in 2D or 3D SMLM experiments respectively
SpheroRuler beads are coated with 647-fluorophores giving a stable blinking in SMLM microscopy, and have been initially developed for dSTORM imaging. Since then, they have also been successfully used in SFFR, SEM, confocal, Airyscan confocal and SEM microscopy.
The spherical particles making up Spheroruler beads have been selected based on very good monodispersity properties. The accuracy and reproducibility of the bead diameter have been characterized by SEM on 25 independent microspheres and showed a standard deviation of 1 +/- 0.05μm.
The SpheroRuler kit contains a 50μL suspension of SpheroRuler beads. All you need to have on your side is some blinking buffer, coverslips and imaging vessels of your choice.
One kit contains a 50μL suspension of SpheroRuler beads, allowing for 10 experiments when using the recommended 5μL volume in 35mm glass-bottom dishes.
Yes, SpheroRuler beads are resuspended in PBS and can be loaded together with biological specimens (cells, tissue sections, etc).
Yes, SpheroRuler beads are resuspended in PBS and can be loaded together with biological specimens (cells, tissue sections, etc).
The SpheroRuler suspension is stable for at least 7 months when stored at 4°C.
In 2D, you should retrieve a hollow circle of 1μm diameter. In 3D, you should retrieve a hollow sphere of 1μm diameter.
SpheroRuler beads are highly fluorescent beads coated with a high density of fluorophores. We recommend using a “multi-emitter / high density” rather than a “single-emitter / low density”, algorithm, to guarantee an efficient localization of individual blinking events. Although the available options will vary on each imaging system used, an example of algorithm that was successfully used is the “Account for overlap” function on the ZEN software (Zeiss).
Fluorophores are directly coated on the bead surface without linkers or antibodies, and their thickness is therefore negligible compared to the measured diameter. The apparent thickness of the fluorophore ring will depend on the resolution of your imaging system (i.e. around 160-200nm when measured in dSTORM). The external periphery of the beads should be taken into account when measuring diameter.
Although 2D reconstructions of SpheroRuler beads should be accurately circular, obtaining an elongated shape in z is a common artefact that will depend on the system used for imaging and for 3D reconstruction (biplane reconstruction, astigmatism, etc). The measured diameter in z and its distance from the actual 1μm can therefore be used as a robust indicator to evaluate the fidelity of the 3D reconstruction for a given system. As an example, measured z diameter was 1.3 μm when tested on a Vutara VXL system using biplane imaging.
The SpheroRuler beads are coated with a high density of fluorophores. A few tips that will facilitate the SpheroRuler reconstruction:
– Make sure you perform a “pumping” step by illuminating with ultra-high power (i.e. laser 80% for less than 30s) before starting the acquisition in order to improve the localization precision.
– Use a “multi-emitter / high density” algorithm, or any equivalent, to guarantee an efficient localization of individual blinking events.
– If still not sufficient to efficiently reconstruct the spheres, or if such algorithm is not available, using a PSF filter to eliminate out-of-focus events or close double emitters should refine the localisation precision and make the peripheral crown more visible, therefore improving the reconstruction fidelity.
Photoinducible protein activator (powder)
A technology developed by Ludovic Jullien, Isabelle Aujard and Thomas Le Saux – ENS Paris, France
| Size | Catalog Number |
| 5 mg | LJU-CIN-5 |
| 50 mg | LJU-CIN-50 |
Actiflash is a small steroid ligand that activates engineered proteins upon light induction. It can be used to precisely control protein activity down to the single cell level in live cell cultures and animals.
")
Actiflash is a tamoxifen-like caged-inducer called cyclofen. Upon UV-light illumination, optical-induced uncaging of Actiflash quickly releases proteins fused to the modified estrogen receptor ligand-binding domains ERT2. This allows them to translocate into the nucleus, activate transcription (using Gal4-UAS system) or induce recombination (using the Cre-lox one).
The protein expression can thus be simply controlled in time and space thanks to UV-light!

Technology capitalizing on the versatile use of Tamoxifen-OH for controlling functions of multiple types of proteins.
Caged Cyclofen-OH liberates Cyclofen-OH, a highly photostable compound in contrast to Tamoxifen-OH encountering photodegradation under illumination.
Uncaging requires either UV-A light or a strong IR laser. Visible light is inactive, which facilitates the experiments with biological samples.
Caged Cyclofen-OH is cell-permeant and can be added either in the external medium or directly injected for conditioning.
Caged Cyclofen-OH does not generate any basal activation of protein function and it benefits from an excellent temporal resolution upon uncaging.
Actiflash is a great R&D tool to convert your inducible ERT model into a photo-inducible one. It is very helpful if you want to control transcription (using Gal4-UAS) or induce recombination (using Cre-lox) in space and/or time for in-vivo cell tracking experiments and more.
Actiflash has been successfully used with various cell lines as well as zebrafish embryos. Actiflash will work best on zebrafish embryos from 12 to 48hpf.
Find below a plasmid list that can be used with Actiflash. All these plasmids are available from Addgene.
Lifetime: upon receipt, Actiflash is stable for years at 2-8°C. Once solubilized in a DMSO solution (typically at 10 mM), Actiflash can be stored at -20°C for several months. Always wrap the vials containing the aliquoted Actiflash with aluminium foil to protect it from light.
It is advised to first establish the extent of phenotype sought for as a function of the Tamoxifen-OH concentration. Then the concentration of Actiflash used for sample conditioning is fixed at Tamoxifen-OH concentration causing 100% of the desired phenotype (in general 3-5 μM in cultured cells and zebrafish embryos).
Incubate your samples in a serum-free medium for 90 mins, away from light.
Illumination of Actiflash may be performed with UV (325-425nm range) light or multiphoton excitation (at 750 and 1064 nm with two- and three-photon excitation, respectively) to release Cyclofen-OH. You can use either benchtop UV lamps or light sources installed onmicroscopes.
The objective is to provide enough photons to exhaust the conversion of the Actiflash but without generating detrimental side-effects on the biological sample.Simply analyze the phenotype recovery with decreasing illumination duration. Then determine the shortest illumination duration leading to 100% uncaging of Actiflash.
Actiflash has been successfully used with various cell lines as well as animal models:
● Cell lines: CV1, mammalian epithelial cells (MDCK)
● Zebrafish: 12 hpf – 2 dpf embryos
● Mice
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
Actiflash works optimally on embryos below 2 dpf when following the provided protocol of use. On older embryos, protocol adjustments might need to be implemented to achieve a satisfactory photoconversion (i.e. heart injection, electroporation, etc).
You will find below a non-exhaustive list of proteins that have been fused to the estrogen receptor:
● Recombinase Cre
● Tyrosine-protein kinase ABL1
● Cellular tumor antigen p53
● Proto-oncogene c-Fos
● Endothelial transcription factor GATA-2
● CMP-N-acetylneuraminate-beta-galactosamide-alpha-2,3-sialyltransferase4 (GAL4)
Actiflash is provided as a powder. It is stable for years at 2-8°C. Once solubilized in a DMSO solution (typically at 10 mM), Actiflash can be stored at -20°C for several months. Always wrap the vials containing the aliquoted Actiflash with aluminium foil to protect it from light.
You can perform experiments with Tamoxifen-OH as a positive control.
After conditioning your sample at a given concentration (e.g. 5 μM) with Actiflash,
illuminate your samples with the shortest illumination duration leading to the maximum
phenotype recovery. Compare with the results obtained with Tamoxifen-OH (1 μM).
Any plasmid containing the ERT sequence should work.
Please find below a list of 52 plasmids developed by Sophie Vriz and Michel Volovitch in The Sophie Vriz Lab, who have the ownership of citations related to these plasmids. These 52 plasmids are all available on Addgene
| Type | Addgene | Vriz lab name & link to Addgene | Coding sequence | Main use | Regulatory sequences |
| ERT2 ready for fusion | |||||
| 184057 | pCSnERT(#186) | nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| ERT2 fused to CRE | |||||
| 184058 | pCSCRET2(#323) | 6myc-CRE-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184059 | pT2EFCRET(#324) | CRE-ERT2 | Zebrafish Expression | X.laevis-EF1 | |
| 184060 | pT2h70CRET(#325) | CRE-ERT2 | Zebrafish Expression | D.rerio-hsp70, SP6 | |
| 184061 | pT22iU66CriT(#387) | CRE-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi, SP6 | |
| 184062 | pT22iD36CriT(#388) | CRE-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Dio3 | |
| ERT2 fused to Fluorescent Protein or Fluorogen-activated peptide | |||||
| 184063 | pCSmChnERT(#99) | mCherry-nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184064 | pCSKaedenERT(#100) | Kaede-nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184065 | pCSGFPnERT(#205) | eGFP-nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184066 | pT22UbGfpnE4PCh(#301) | eGFP-nls-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184067 | pT2iC6Dronpa2nlsER4(#636) | Dronpa2-2nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184068 | pT2iC6FAST1nls2ER4(#638) | YFAST-2nls-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| ERT2 fused to Gal4 activator | |||||
| 184069 | pCSTG4FER3(#190) | Gal4bdFF-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184070 | pT22gCfpUbiG4FER4P2A(#249) | Gal4bdFF-ERT2-P2A | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184071 | pT2iaTubG4FER3(#940) | Gal4bdFF-ERT2 | Zebrafish Expression | C.auratus-a1Tub | |
| 184072 | pT22i2Shh24G4FER3(#1041) | Gal4bdFF-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Shha+ABC | |
| 184073 | pT26CfpSox10G4FER3(#1048) | Gal4bdFF-ERT2 | Zebrafish Expression, stable transgenesis (blue heart) | D.rerio-Sox10 | |
| 184074 | pT26i4Gli8G4E3(#1254) | Gal4bdFF-ERT2 | Zebrafish Expression, stable transgenesis (blue heart) | D.rerio-GliRE | |
| ERT2 fused to various kinases, inhibitors, redox enzymes | |||||
| 184075 | pCSZPKIaHAERT(#118) | ZfPKAinhiba-HA-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184076 | pCSZPKIamutHAERT(#119) | ZfmutPKAinhiba-HA-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184077 | pCSmp38ERT(#143) | 5myc-ZfP38a-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184078 | pT22U6CatalE4PCh(#767) | CatalDeltaC-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi, SP6 | |
| ERT2 fused to various vertebrate transcription factors | |||||
| 184079 | pCSmEn2ERT(#117) | 6his-myc-chkEn2-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184080 | pCSmEn2SRERT(#231) | 6his-myc-chkEn2(SR)-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184081 | pCSmEn25EERT(#232) | 6his-myc-chkEn2(5E)-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184082 | pCSmEn2n4ERT(#234) | 6his-myc-chkEn2(n4)-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184083 | pT22gCfpUbm5En2E4PCh(#251) | 5myc-chkEn2-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184084 | pT22UbHEn2E4PCh(#262) | 3HA-chkEn2-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184085 | pT22UbhaEng2aE4PCh(#271) | 3HA-ZfEng2a-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184086 | pT22UbhaEng2bE4PCh(#272) | 3HA-ZfEng2b-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184087 | pT22Ubm5Eng2aE4PCh(#283) | 5myc-ZfEng2a-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184088 | pT22Ubm5Eng2bE4PCh(#284) | 5myc-ZfEng2b-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184089 | pT22Ubm5En2SRE4PCh(#302) | 5myc-chkEn2(SR)-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184090 | pCSm5En2CER4(#457) | 5myc-chkEn2(C>S)-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184091 | pT22Ubm4Eng2bKRPC(#493) | 4myc-ZfEng2b(WW>KK)-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184092 | pT22gcfUbm5En25EE4PCh(#552) | 5myc-chkEn2(5E)-ERT2-P2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184093 | pCSm5En2DER4(#1062) | 5myc-chkEn2(C>D)-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184094 | pT22cUm5En2E4P3Che(#1188) | 5myc-chkEn2-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184095 | pT22cUm5En2SE4P3Che(#1189) | 5myc-chkEn2(C>S)-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184096 | pT22i5uasm5En2E4P3Che(#1208) | 5myc-chkEn2-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | 5xUAS, SP6 | |
| 184097 | pT22i5uasm5En2SE4P3Che(#1209) | 5myc-chkEn2(C>S)-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | 5xUAS, SP6 | |
| 184098 | pT22i5uasH3En2SE4P3Che(#1212) | 3HA-chkEn2(C>S)-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | 5xUAS, SP6 | |
| 184099 | pT22i5uasm5En25EE4P3Che(#1213) | 5myc-chkEn2(5E)-ERT2-5’fP2A-mCherry | Zebrafish Expression, stable transgenesis (blue eye) | 5xUAS, SP6 | |
| 184100 | pT2iD6Hoxa13aER4(#685) | ZfHoxa13a-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184101 | pT2iD6Hoxa13bER4(#686) | ZfHoxa13b-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| 184102 | pT2iU6m5Hoxa13bE4PCh(#765) | 5myc-ZfHoxa13b-ERT2-P2A-mCherry | Zebrafish Expression | D.rerio-Ubi, SP6 | |
| 184103 | pCSThaP6ERT(#184) | HA-MmPax6-ERT2 | Mammalian Expression, mRNA in vitro synthesis | sCMV, SP6 | |
| ERT2 fused to Caspase-9 (apoptosis actuators and sensors) | |||||
| 184104 | pT2iApowriter2(#336) | myr-DIAP1-5myc-tEosFP-nls’-P2A-Casp9-ERT2 | Zebrafish Expression | D.rerio-Ubi | |
| 184105 | pT22Apowriter2(#340) | myr-DIAP1-5myc-tEosFP-nls’-P2A-Casp9-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | D.rerio-Ubi | |
| 184106 | pT2iU6Apowriter2N(#355) | myr-DIAP1-5myc-tEosFP-nls’-P2A-Casp9-ERT2 | Zebrafish Expression | D.rerio-Ubi, SP6 | |
| 184107 | pT22iuasvApoWtEos3N(#486) | myr-DIAP1-2nls-tEosFP-nls’-P2A-Casp9-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | 4xUAS | |
| 184108 | pT22iuasvApoWChe(#488) | myr-DIAP1-nls’-mCherry-P2A-Casp9-ERT2 | Zebrafish Expression, stable transgenesis (blue eye) | 4xUAS |
With the Actiflash conditioned samples, analyze the phenotype recovery observed using a
100% laser power and a decreasing illumination duration. You will thus determine the
shortest illumination duration leading to 100% uncaging.
Examples of the duration required for uncaging 100% of Actiflash:
• One-photon excitation at a 365 nm wavelength with a benchtop UV lamp having a power
range of 4-6 W (e.g. for global photoactivation): 5 minutes.
• One-photon excitation in the 350-405 nm, laser excitation power P = 10 μW focused on a
spot of diameter 10 μm: a few seconds.
• Two-photon excitation delivering 200 fs pulses at 750 nm and P = 10 mW in a cell of volume
V =100–1000 μm3: 1 second.
Actiflash has been designed by Ludovic Jullien , Isabelle Aujard and Thomas Le Saux
Additional publications:
Feng, Z., Ducos, B., Scerbo, P., Aujard, I., Jullien, L., & Bensimon, D. (2022). The Development and Application of Opto-Chemical Tools in the Zebrafish. Molecules (Basel, Switzerland), 27(19), 6231. https://doi.org/10.3390/molecules27196231
Ready-to-use silicon chambers to stick and remove for cellular confinement or dynamic assays, such as migration or wound-healing.
Each product contents 100 stencils (2 sheets of 50 stencils)
")

Ready-to-use silicon chambers to stick and remove for cellular confinement or dynamic assays, such as migration or wound-healing.
Each product contents 100 stencils (2 sheets of 50 stencils)
| Description | Catalog Number |
| Solo : 1 circular well – Diam. 12mm | STU-STE-1WL |
| Quartet : 4 circular wells – Diam. 3mm | STU-STE-4WL |
| Nonet : 9 circular wells – Diam. 3mm | STU-STE-9WL |
| Allegro : 2 oblong wells spaced by 0.50 mm | STU-STE-LRG |
| Presto : 2 oblong wells spaced by 0.25 mm | STU-STE-THN |
Stencell are ready-to-use silicon chambers that you can stick and remove for cellular confinement or dynamic assays, such as migration or wound-healing.
Stencell was designed to standardize your experiments and minimize the consumption of reagents.
It is very helpful when you are working with super expensive reagents or very rare cell lines, and when you want to test various experimental hypothesis.
Cell migration and wound healing with Presto and Allegro
Parallelized experiments with Quartet and Nonet
Confined experiments with Solo

Material: PDMS
Kit content: 100 stencils (2 sheets of 50 identical stencils)
Sizes & shapes:
– Solo: 1 circular well – Diam. 12 mm
– Quartet: 4 circular wells – Diam. 3 mm
– Nonet: 9 circular wells – Diam. 3 mm
– Presto: 2 oblong wells spaced by 0.25 mm
– Allegro: 2 oblong wells spaced by 0.50 mm
Storage: up to 2 years, at room temperature under their protective sheets.
Stencell was designed by Vincent Studer, Pierre-Olivier Strale and Aurélien Pasturel.
They were hosted by the Cell Organ-izers joint research laboratory (CNRS-Alvéole).
Original publication:
Pasturel, A., Strale, P. O., & Studer, V. (2020). Tailoring Common Hydrogels into 3D Cell Culture Templates. Advanced healthcare materials, 9(18), e2000519. https://doi.org/10.1002/adhm.202000519
The device to confine and study your cell behavior within a physiological rigidity range.
Kit contents:
. 1 Agarsqueezer device
. 2 needles

Silicium wafers to mold confining pillars of various heights can be bought separately here.
A technology developed by Audrey Prunet, Gilles Simon, Hélène Delanoë-Ayari, Véronique Maguer-Satta & Charlotte Rivière – ILM Lyon, France
AgarSqueezer is a microscope slide chamber equipped with a molded agar-based compression system. Use it to apply an instant & homogeneous compression on your cells, and study their response to short and long-term confinement.

Contrary to PDMS, the mechanical properties of agarose reproduce stiffness of the in vivo microenvironment (1-150 kPa).
As the porous nature of agarose facilitates nutrient and oxygen diffusion, the AgarSqueezer provides a safe environment for long-term cell culture. Keep your cells confined for up to 10 days!
By modulating confinement level, matrix stiffness & composition or coating with ECM proteins, AgarSqueezer leaves you plenty of options to reproduce environmental cues.
Agarsqueezer has been successfully used to confine adherent and non-adherent cells, including:
> Human: primary T-lymphocytes, TF1 & ML2 leukemic cells, HS27A fibroblasts, MCF10A breast cells, MDA-MB-231 breast cancer cells, U-2 OS osteosarcoma cells, PC-3 & DU 145 prostate cancer cells, HT29 & HCT116 colorectal adenocarcinoma cells and HT1080 fibrosarcoma cells
> Murine: megakaryocytes, osteocyte-like cells MLO-Y4, primary muscle cells & primary dendritic cells
> Plant: cells from Arabidopsis roots
> 3D cell cultures: mice gastruloïds
Compatible assays:
> Live imaging*: cell viability, proliferation, migration, morphology, nuclear deformability, cytoskeleton reorganization, cell tracking, etc
> Standard molecular analysis (immunostaining, western blot, qPCR, flow cytometry, etc): changes in gene/protein expression, proliferation, morphological analyses, etc
*Cells can be added with fluorescent probes or stimulated with drugs during confinement.
Imaging modes:
Inverted microscopes equipped with a standard adjustable petri dish holder, with any imaging mode: phase-contrast, epifluorescence, confocal, etc.
1x Agarsqueezer compression device
1x 16G flat cut needle to make holes in the agar gel, facilitating diffusion of culture medium or drugs during the experiment
1x 20G flat cut needle (same as above)
(Optional) 1x microscope stage insert (102.5mmx143.5mm), holding up to 2 AgarSqueezers in live imaging chambers (Okolab or equivalent).
In addition to basic kit contents, we recommend using a silicium wafer to mold your confining pillars in agarose. We provide four different wafers to mold pillars of various heights. Choose among our four pillar heights available depending on your desired application and add silicium wafer(s) to your order here.
")
")
Quantification of cell morphology under confinement. (A–C): Morphology of immature TF1-GFP hematopoietic cells for control (A) and for 30 μm and 5 μm (B and C, respectively). Scale bar = 20 μm. (D) Quantification of projected area by automatic image analysis. Projected area was similar for control and 30 μm, while it significantly increased for 5 μm confinement (unpaired t-test n = 160 at least for each condition). (E and F): z-Section of unconfined (E) and confined (F) cells. Scale bar = 10 μm.
et al. Lab on Chip, 2020.
Human T-lymphocytes isolated from blood were seeded in the Agarsqueezer and confined under 5µm pillars. Images were taken every 10sec for 16.5min using an inverted Zeiss Z1 automated microscope (10X objective).
Credits: Marie-Pierre Valignat – Adhesion & inflammation lab, Aix-Marseille University, France

Arabidopsis thaliana Col-0 root cells stained with Calcofluor (cell wall) and imaged with a confocal microscope either in a traditional culture setting (left), or after 24h of confinement in the Agarsqueezer using the 30µm (middle) or 5µm (right) pillars.
Credits: Léa Bogdziewiez – UPSC – Sveriges lantbruksuniversitet, Sweden

C57 primary myoblasts stained with Hoechst were imaged in the AgarSqueezer before compression (left panel), and after 1.5h of compression under 2.5 µm height pillars (middle & right panels).
Credits: Dr. Hind Zahr & Dr. Alice Varlet, Lammerding Lab – Meinig School of Biomedical Engineering, Cornell University, United States

")
")
The Agarsqueezer device has been successfully used to study compression of adherent and
non-adherent cells, including:
Human cells:
Fibrosarcoma (HT-1080)
Osteosarcoma (U-2 OS)
Colorectal adenocarcinoma
(HT29 & HCT116)
Prostate cancer (PC-3 & DU 145)
Breast cancer (MDA-MB-231)
Leukemia (TF1 & ML2)
Megakaryocytes
Fibroblasts (HS27A)
Breast cells (MCF10A)
Primary T-lymphocytes
Murine cells:
Osteocyte-like cells (MLO-Y4)
Primary dendritic cells
Primary muscle cells
Plant cells:
Arabidopsis Thaliana root cells
3D cell culture:
Mice gastruloids
The system is fully compatible with live imaging and time-lapse microscopy and all
immunostaining steps can be performed in situ. Alternatively, cells can be collected for
classical biochemical and molecular analysis (qPCR, Western Blot, flow cytometry, etc).
The device has been designed for long-term confinement studies and the porous nature of
agarose enables passive medium renewal as well as oxygen diffusion. You can safely grow cells
up to 10 days in the AgarSqueezer device.
Yes, the use of agarose allows free diffusion of small molecules (size <30 nm in 2% agarose).
You can easily add drugs, antibodies or other compounds at any point during cell confinement
studies thanks to an open access to the reservoir. Drug availability and activity within the
system has been validated using in situ addition of the tyrosine kinase inhibitor imatinib, and
diffusion experiments showed that 3 hours are required for the diffusion of small molecules
such as BSA or FITC.
The currently available pillar heights are:
– 2.5μm for inducing highly confined conditions
– 5μm for inducing moderately confined conditions
– 30μm to serve as an unconfined control
– 100μm to confine 3D cellular structures or immobilize them for imaging
Those dimensions have been determined based on the size of most classical human cell lines.
The resulting level of compression applied will depend on your sample type, size and the use
of mono- or multi-cell layers.
You can easily modulate the matrix stiffness by tuning the type and concentration of agarose used to mold the pillars. Using a standard agarose at the recommended 2% concentration will result in a storage modulus within the range of 50-200kPa. To accentuate the matrix stiffness and the resulting confinement applied, the agarose concentration can be increased. For example, a 3% agarose was found to be more efficient at confining cell cultures from Arabidopsis roots. If you wish to decrease the matrix stiffness, we recommend using a “low
melting point” or “ultra-low gelling” temperature agarose to yield a storage modulus in the range of ~1kPa. For a similar stiffness on top and bottom walls, the coverslip can be coated with an additional soft agarose layer.
To analyze the role of various ECM proteins on cell response to mechanical confinement, you
can coat the coverslip with adhesive proteins (fibronectin, collagen, Matrigel, etc). In addition,
the agarose can be added with collagen, PEGDA with covalently immobilized RGD peptides or
silk.
Our AgarSqueezer kits contain all elements making up the compression device, a wafer to mold your agarose gel and some needles specifically designed to form your entry ports in the agarose. Optionally, an insert (102.5mmx143.5mm) designed to image 2 AgarSqueezer devices in parallel can be added to your order. This insert will fit into Okolab imaging chambers or equivalents.
All you need to have on your side is:
• Agarose powder
• Distilled water
• Round 22 or 30 mm coverslips
• A screwdriver
• For imaging: a standard petri dish holder can be used to place and image the AgarSqueezer on inverted microscopes. Use objectives up to 40X for optimal performance. Although objectives of 60X and above can be used, they will result in a shorter observation area and we cannot guarantee an optimal working distance depending on your microscope.
Yes, the AgarSqueezer device can be re-used as long as it is thoroughly sterilized by autoclaving
before each experiment. Simply keep in mind that the agarose solution should be prepared
no more than one day before your experiment.
Agarsqueezer has been designed by Audrey Prunet, Gilles Simon, Hélène Delanoë-Ayari, Véronique Maguer-Satta and Charlotte Rivière (ILM Lyon, France).
Additional publications:
Mitosis down-regulates nuclear volume and resets nuclear envelope folding of cancer cells under prolonged confinement
Malèke Mouelhi, Alexis Saffon, Hélène Delanoë-Ayari, Sylvain Monnier, Charlotte Rivière, Biorxiv 2023
doi: https://doi.org/10.1101/2023.05.11.540326
Mitosis sets nuclear homeostasis of cancer cells under confinement
Maleke Mouelhi, Alexis Saffon, Morgane Roinard, Helene Delanoe-Ayari, Sylvain Monnier, Charlotte Riviere, Biorxiv 2023. doi: https://doi.org/10.1101/2023.05.11.540326
A stamp to mold agarose wells to culture and image spheroids and organoids
")
| Size | Catalog Number |
| V300 | GRE-MOU-V300 |
| V500 | GRE-MOU-V500 |
Stampwell stamps imprint arrays of wells in hydrogels in just one time. Nothing more to do than load your samples into the wells and image. The Stampwell shapes have been optimized for the imaging of spheroids and organoids.
V Shape 300µ:
. Number of pins: 42
. Depth of the well: 1 mm
. Well bottom diameter: 300µm – Shape: circular
. Well upper side: 1mm*0.5mm – Shape: rectangular


V Shape 500µ:
. Number of pins: 42
. Shape of the pins: V
. Depth of the well: 1 mm
. Well bottom diameter: 500µm – Shape: circular
. Well upper side: 1mm*0.5mm – Shape: rectangular

")
Soaked in fluorescein, negative rendering
Image credit: Gaëlle Recher – Bordeaux
scale bar: 500µm

Image credit: Gaëlle Recher – Bordeaux
Notice that the focus moving through the well height is easily visible (lines in focus Vs out of focus)
Image credit: Gaëlle Recher – Bordeaux
Filled with a single cell-loaded alginate capsule: corresponding imaged with a stereomicroscope and stitched. Gel pad is made of 2% agarose. (Scale bar: 2mm)
")
Image credit: Gaëlle Recher – Bordeaux

Airway organoids were generated from a human bronchial cell line in hydrogel and transferred either to microwells made using a V-shape Stampwell, or suspension plates, for turning them inside out and imaging.
Credits: Signe Lolle – Danmarks Tekniske Universitet, Denmark

1. Pour liquid agarose (or other hydrogel)
2. Place the stamp
3. Reticulate the hydrogel
4. Remove the stampv
Original publication:
Stampwell has been originally developed by Gaelle Recher and published in Scientific Reports:
Alessandri K, Andrique L, Feyeux M, Bikfalvi A, Nassoy P, Recher G. All-in-one 3D printed microscopy chamber for multidimensional imaging, the UniverSlide. Sci Rep. 2017 Feb 10;7:42378. doi: 10.1038/srep42378. PMID: 28186188; PMCID: PMC5301227.

Mold agarose wells to position and image zebrafish larvae and embryos
| Size | Catalog Number |
| Embryo 1 | GRE-MOU-REC |
| Embryo 2 | GRE-MOU-LAI |
| Larvae 1 | GRE-MOU-DEV |
| Larvae 2 | GRE-MOU-ADU |
Stampwell stamps imprint arrays of wells in hydrogels in just one time. Nothing more to do than load your samples into the wells and image. The Stampwell shapes have been optimized for the imaging of fish embryos or larvae up to 20 dpf.
Save time: no need to manually position your samples individually. Fish embryos/larvae will self-position in the wells.
Boost your assay reproducibility: once loaded in the wells, your samples are perfectly positioned, aligned and located on a similar focal plane.
Open system: direct access to embryos/larvae for adding external compounds (i.e. drug screening).


Embryo 1 (ie Rectangular)
. Number of pins: 35
. Shape of the pins: rectangular
. Length * width of the wells: 2 mm * 0,65 mm


Embryo 2:
. Number of pins: 18
. Shape of the pins: a drop
. Length of the wells: 3.90 mm
. Largest width of the wells: 0.88 mm


Larvae 1:
. Number of pins: 5 large pins ideal for the 9-14 dpf zebrafishes + 5 medium pins ideal for the 6-9 dpf zebrafishes + 5 small pins ideal for the 3-6 dpf zebrafishes
. Shape of the pins: fish body and tail
. Length of the 5 large wells: 6 mm, 2.7 for the body and 3.3 for the tail
. Length of the 5 medium wells: 5 mm, 2.25 for the body and 2.75 for the tail
. Length of the 5 small wells: 4 mm, 1.8 for the body and 2.2 for the tail


Larvae 2:
. Number of pins: 5 large wells ideal for the 15-20 dpf zebrafish or 39-42 dpf medaka larvae + 5 small wells ideal for the 1-2 dpf zebrafish
. Shape of the pins: prism
. Length of the 5 large wells: 10 mm
. Length of the 5 small wells: 3 mm
. Well depth: 1 mm

48hpf embryos (anti-HuC/D, ab210554, abcam)
Credits: Matthieu Simion, CNRS, 2022

Credits: Dr Rui Monteiro – Institute of Cancer and Genomic Sciences, University of Birmingham, United Kingdom

Bar: 1mm (projection of Z-stack – confocal microscope)
Credits: Gaëlle Recher – Bordeaux 2019

Top picture: green-heart 3dpf zebrafish larvae placed in ventral view. Middle & bottom pictures: 3dpf zebrafish larvae PFA-fixed,
mounted in glycerol and inserted in Larvae 1 Stampwell (anterior to the left). Pictures were taken using a Leica Spectral Confocal SP5.
Credits: Dr. Giovanni Risato, Prof. Natascia Tiso and Alina Ramazanova – Department of Biology, University of Padova, Italy
Anesthetized 3 dpf embryos expressing a fluorescent biosensor in the heart were mounted in agarose microwells made using the Larvae 1 Stampwell and imaged in ventral view.
Credits: Prof. Juan Llopis – CRIB, Universidad de Vastilla La Mancha, Spai
1. Pour liquid agarose (or other hydrogel)
2. Place the stamp
3. Reticulate the hydrogel
4. Remove the stampv
Original publication:
Stampwell has been originally developed by Gaelle Recher and published in Scientific Reports:
Alessandri K, Andrique L, Feyeux M, Bikfalvi A, Nassoy P, Recher G. All-in-one 3D printed microscopy chamber for multidimensional imaging, the UniverSlide. Sci Rep. 2017 Feb 10;7:42378. doi: 10.1038/srep42378. PMID: 28186188; PMCID: PMC5301227.
SensiDeath is a novel sensitive kit for human cell death detection. Taking benefit from qPCR high sensitivity, SensiDeath provides a simple method to detect cell death at its earliest stages by quantifying genomic DNA fragments released in the cytoplasm. Use it to get reliable measurements of apoptosis across a range of cell populations.
| Product | Catalog No. |
| SensiDeath – Ultra-sensitive human cell death assay -300 assays | IDY-SD-300-IDY |
| SensiDeath – Ultra-sensitive human cell death assay – 1000 assays | IDY-SD-1000-IDY |
Fast & simple
As simple as a qPCR. Lyse your cells, collect the cytosolic fraction and get results in less than 2h !
Extended dynamic range
Get reliable data and make sure to never miss out subtle changes in cell death.
Scalable
Compatible with 96-well plate format and efficient on small cell populations, SensiDeath is a convenient method for high-throughput cytotoxicity assays.
Early detection
Based on extremely sensitive cytosolic detection of genomic DNA, SensiDeath detects cell death as soon as it is initiated and up to its late phase.
SensiDeath is based on a patented technology relying on the quantitation of genomic DNA released in the cell cytosol, a key process initiated upon cell death and a valuable marker of apoptosis. By amplifying genomic DNA present in isolated cytosolic fractions, you can quickly compare relative levels of apoptotic cells across a wide range of samples.
Outputs: comparative measurements of cell death levels for cytotoxic or viability assays. SensiDeath can be used as a reliable method to distinguish changes in apoptosis*.
*Check out our Results section for detailed results.
Sample types: human cells (adherent & suspension)*.
*Check out our FAQ section for a full list of validated cell lines.
Analysis: PCR-based techniques (quantitative PCR, digital droplet PCR).
All our kits include a pair of optimized primers designed to amplify a genomic repeat DNA element. They have been thoroughly selected and optimized to combine high sensitivity, specificity and reproducibility in cell death detection.
Our kits also include a concentrated lysis buffer, developed and optimized to recover cytosolic fractions by disrupting the cell plasma membrane specifically while maintaining integrity of the nuclear membrane as well as that of other cellular organelles.
Small kit (>300 assays*)
Primer mix (20X): 300 µL
SensiDeath Lysis buffer (6X): 25 mL
Extended kit (>1000 assays*)
Primer mix (20X): 1 mL
SensiDeath Lysis buffer (6X): 85 mL
*Number of assays are given for 20 µL qPCR reactions. More assays can be carried out with each kit if working with smaller qPCR reaction volumes.
Storage: the 6X lysis buffer can be stored at 4°C for at least 1 year. Primers can be stored at 4°C for one month, or at -20°C for long-term storage.

Caspase-Glo® 3/7 or SensiDeath kits were used to measure cell death in increasing quantities of MOLM14 cells treated with 1 µM Etoposide for 16 hours. SensiDeath provides a 4-fold increase in detected signal compared to Caspase-Glo® 3/7 in treated samples and shows a lower detection limit.
Image credits: Equipe ANUBIS, Université de Toulouse, France

Annexin-V or SensiDeath methods were used to measure cell death in MOLM14 cells treated with increasing concentrations of Etoposide for 16 hours. Compared to Annexin-V, with which a signal upper limit is reached from 2.5 µM Etoposide, SensiDeath provides an extended dynamic range with additional intermediate values available up to 10µM Etoposide.
Image credits: Equipe ANUBIS, Université de Toulouse, France
During cell death, fragmented genomic DNA accumulates in the cytosol. This process takes place as soon as cell death is initiated, and persists up till later stages. Using a selective lysis buffer and qPCR primers, SensiDeath allows a rapid and highly sensitive cell death quantification by detecting genomic DNA fragments released in the cytosol.
Yes, the specific primers provided in the SensiDeath kit are designed to amplify apoptotic DNA generated as soon as cell death is initiated. Several assays confirmed that SensiDeath allowed to distinguish changes in apoptosis using a variety of apoptotic inducers. Check out our Results section for detailed results.
The presence of nuclear DNA in the cytosol can be detected in early, mid and late phase apoptosis. The cytosolic concentration of nuclear DNA will be maximal in the mid-phase, but the extreme sensitivity of the SensiDeath method enables its detection in its earliest phases too.
The SensiDeath method relies on the amplification by qPCR of genomic DNA present in the cell cytosolic fraction. By amplifying repeated genomic sequences present in the cytosolic fraction, you can assess the relative cytosolic enrichment in genomic DNA in comparison to an untreated, negative control. This enrichment will be directly proportional to the cell death levels in your sample.
SensiDeath has been successfully used to detect cell death in a variety of human cell lines, including:
– Adherent cells: HepG2, MDA-MB-231, HeLa, SK-Hep, HEK293, HUVEC, primary fibroblasts, etc
– Suspension cells: OCI-AML3, MOLM14, MV4-11, HL-60, THP-1, U937, etc
Please note that this list is non-exhaustive and SensiDeath could be used to assess cell death in any other human cell types.
SensiDeath is perfectly well-suited for high-throughput and automated assay formats. Extracted cytosolic fractions can be analysed in 96-well or smaller plate formats. The centrifugation step can be replaced by a magnetic-bead system to purify cytosolic DNA fragments.
We provide optimized primers for amplifying a 153 bp genomic repeated element. This primer pair has been meticulously selected to ensure high sensitivity, specificity and reproducibility.
Indeed, some DNA can also be present in the cell cytosol during other processes, especially mitosis. Some tests have been carried out to check whether changes in the cell cycle phases might skew the obtained cytotoxicity data. Non-apoptotic cells were labelled with propidium iodide, and sorted using flow cytometry according to their cell cycle phase (G0/G1, S or G2/M). The SensiDeath protocol has been performed on these samples, and results showed similar qPCR signals across the different samples demonstrating that changes in the cell cycle phase did not affect the obtained data.
The SensiDeath kits contain a concentrated lysis buffer and optimized primers for genomic DNA amplification. The only reagent needed is nuclease-free water and any SYBR Green qPCR mix. In terms of equipment, you need a simple benchtop centrifuge for sample preparation and a thermocycler for performing the qPCR. You do not need any sophisticated equipment.
After lysate centrifugation, nuclear fractions are pelleted at the bottom of the tube. A few microliters of the supernatant are collected for analysis, thus excluding the risk of nuclear contamination.
Yes, nucleases are efficiently inhibited by the lysis buffer. Extracted cytosolic fractions can be kept at -20°C for months before being assayed. This is particularly useful when performing kinetics studies, or simply to save time by assaying multiple samples in parallel.
Yes, the lysis buffer can be added directly to cell culture medium as long as its final concentration is 1X.
Yes, the qPCR amplification signal will reflect the basal level of apoptosis in the control cell population.
No, our lysis buffer provided in the kit has been specifically optimized to disrupt the plasma cell membrane while leaving the nuclear membrane, as well as that of all other organelles, perfectly intact. Therefore, there is no risk of retrieving any mitochondrial DNA in the isolated cytosolic fractions.
The SensiDeath lysis buffer provided in the kit has been specifically developed for destructing the cell plasma membrane while keeping the membrane of nuclei and other organelles intact, therefore allowing the specific retrieval of the cytosolic fraction. In addition, its components have been optimized not to interfere with subsequent qPCR reactions. Therefore, we do not recommend using other lysis buffers as we cannot guarantee the accuracy of the obtained cell death measurements.
Yes, the SensiDeath lysis protocol preserves the integrity of the nucleus and other organelles (mitochondria, ER and Golgi) while extracting the cytosolic fraction. Therefore, it is entirely possible to recover the organelle and nucleoplasm contents to carry out complementary assays from the same lysate. This has been successfully done using detergent-based sequential subcellular fractionation for Western blot analysis.
Red fluorescent probe labeling all mitochondria for live imaging & flow cytometry
GlowMito provides a non-toxic solution to quickly visualize the entire mitochondrial network in live samples.
GlowMito quickly penetrates cells and produces a bright & stable red labeling of mitochondria without inducing cell toxicity or altering mitochondrial functions. Simply add it to your culture medium, and image repeatedly for up to 3 days without washing!
Soluble in water and compatible with 2-photon microscopy, GlowMito is the solution of choice for thick tissues and in vivo experiments.
GlowMito is retained in the mitochondria of live samples following loss of membrane potential.
Colour: Red/near-infrared
Detection method : Fluorescence, one & two-photon excitation
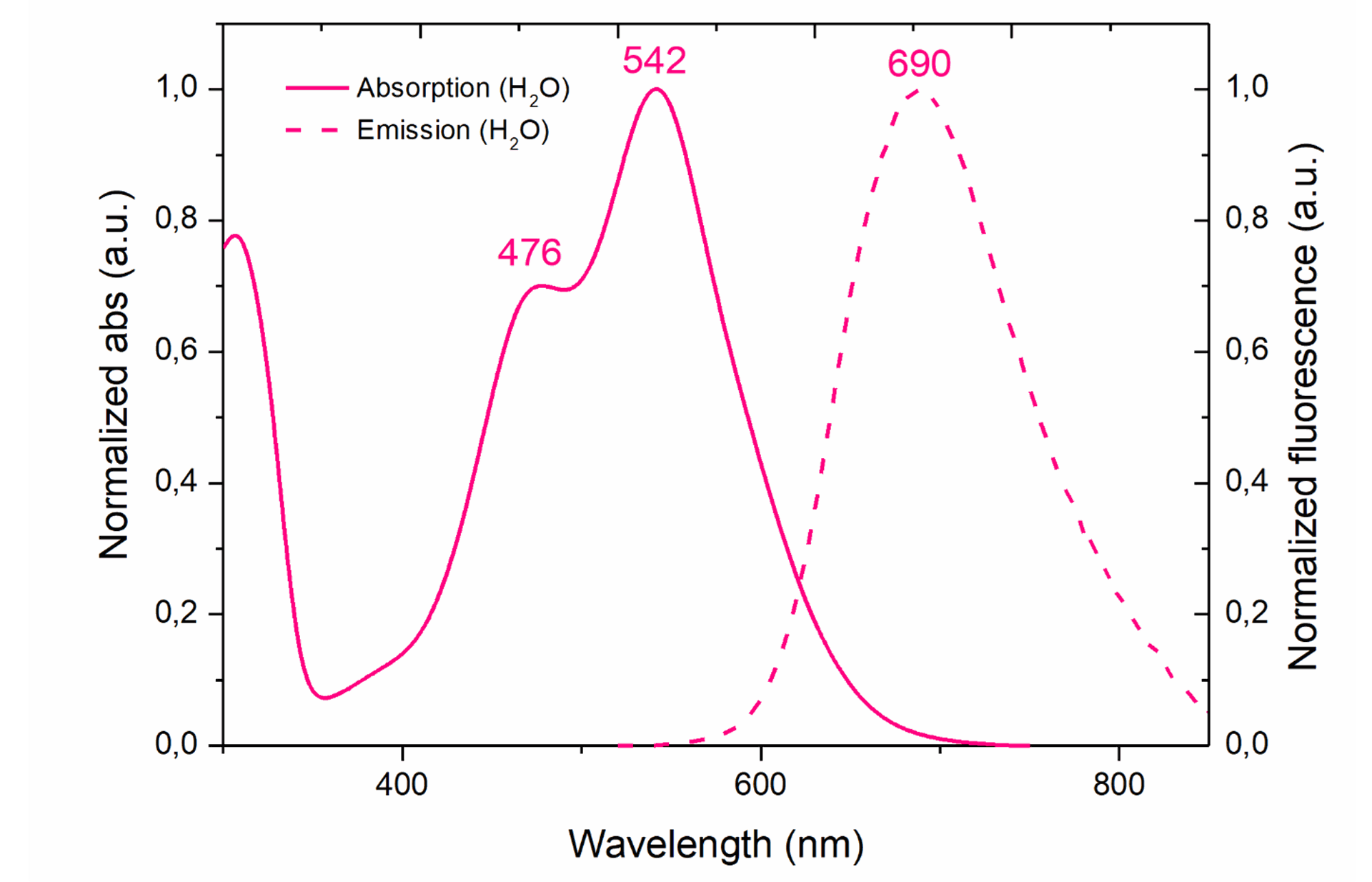
Peak excitation/emission wavelengths: Ex:542nm/Em:690nm
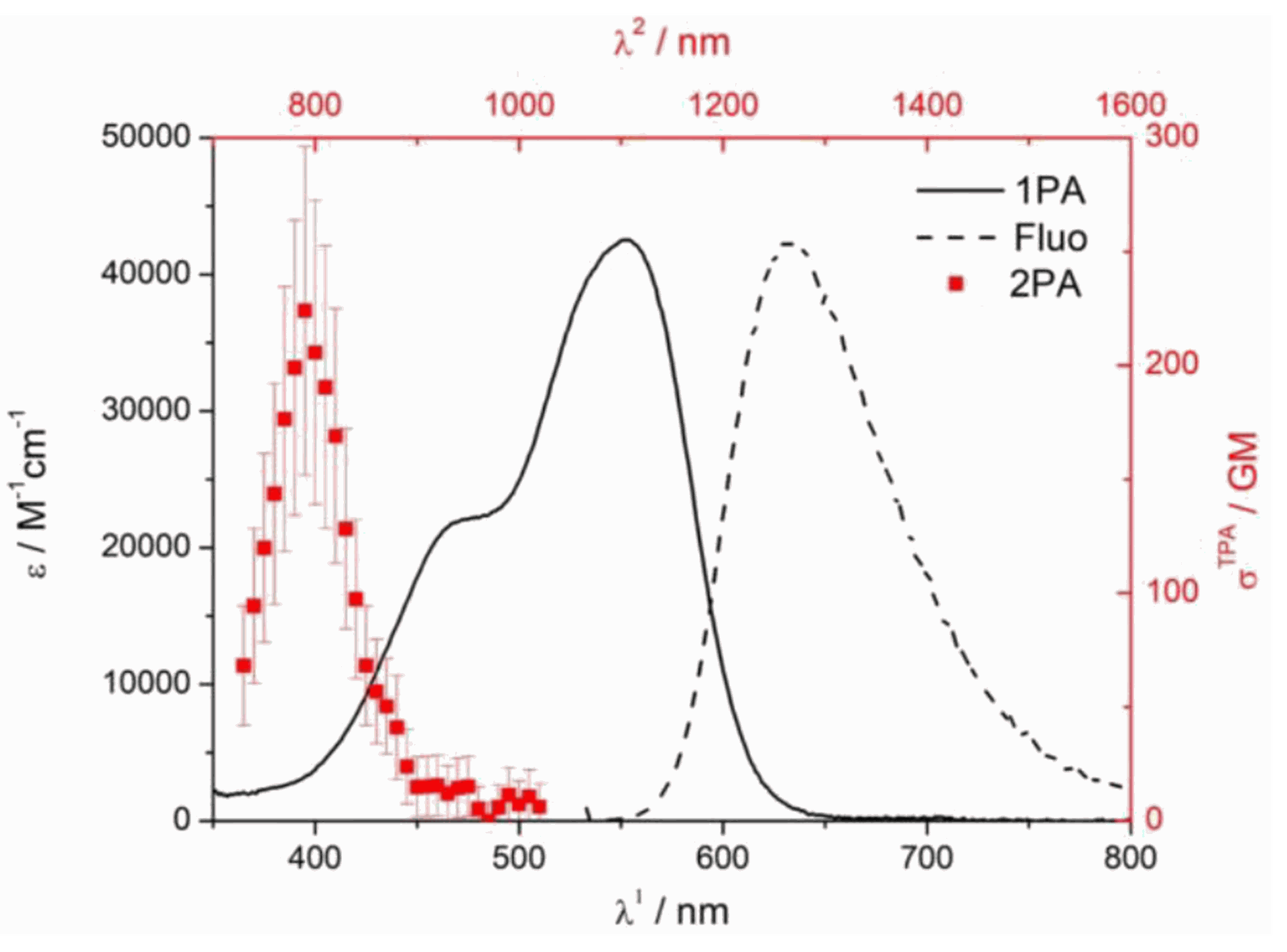
Two-photon absorption peak: 790nm
Two-photon cross-section: 224 GM
Form : Liquid (resuspended in water)
Sub-cellular localization: mitochondria
For use with: epifluorescence & confocal microscopes, flow cytometers
Fixable: no

1 vial contains 125 µL of an aqueous solution with a concentration of 1 mM for a final volume of 250 mL of imaging medium
Storage: Store at 4°C for up to 7 months. For long-term storage, aliquot and store at -20°C.
Experimental outputs: live imaging & flow cytometry
GlowMito produces a bright red labeling of the entire mitochondrial network, including those with reduced potential. It is primarily intended to visualize mitochondria based on their presence rather than assessing their electrochemical properties. GlowMito does not need to be washed out: mitochondrial fluorescence will remain stable for up to 3 days without causing toxicity and can therefore be integrated in your long-term experiments.
GlowMito is perfectly suitable for studying mitochondrial morphology, movement (velocity, localization), tracking, dynamics (fusion/fission), biogenesis (replication & growth), autophagy, etc.
GlowMito can be used in combination with other probes (GFP, potentiometric dyes, calcium signaling probes, etc).
We do not recommend its use for the measure of mitochondrial mass or volume density.
Tested and validated on cell lines, tissues & unicellular organisms:
– Humans: HEK293, HeLa, MCF-7, MDA-MB-231, HMLE, UACC-62, U2-OS, Gli36, HAEC, SH-SY5Y, A-172, A549, patient-derived skeletal muscle cells, primary smooth muscle cells & iPSC-derived cardiomyocytes, hiPSC-derived heart tissues.
– Mice: primary cortical neurons, acute brain slices, isolated pressurized blood vessels
– Monkey: COS-7
– Parasitic protists: Trichomonas Vaginalis (hydrogenosome labeling)
GlowMito showed no internalization in yeast cells, Entamoeba histolytica, Giardia intestinalis parasites and seedlings.
This confocal microscope image depicts the smooth muscle cell layer of an intact pressurized cerebral artery, illustrating mitochondria (shown in magenta) along with the nuclei (shown in blue). Smooth muscle cell nuclei are elongated and run perpendicular to the vessel axis. Small, rounded nuclei correspond to adventitial cells on the vessel wall.
Credits: Image captured by Safa in Dr. Charles Norton’s laboratory – University of Missouri (US)

300 µm thick acute mice brain slices from the primary visual cortex were labeled with 1 µM GlowMito and imaged on a 2-photon microscope (820 nm, 23 mW). Z-stack pictures were taken from top to bottom of the slice (2 µm steps between each image).
Credits: Lauren Panzera (Postdoctoral Fellow Higley Lab) & Michael Higley (Dept. Of Neurosciences) – Yale University, New Haven, CT USA

GlowMito was directly added at 500 nM in live COS-7 culture medium. Cells were imaged repeatedly

HEK293 cells were observed before and after 2, 4, and 6 minutes of incubation with GlowMito 500 nM.
Acquisition parameters: laser 2%, gain 15
A maximum of internalization is observed after 4 minutes.
Credits: Morgane LeMao, Guy Lenaers, Olivier Siri – MitoLab, Unité MitoVasc, Université d’Angers, CNRS UMR6015, Inserm U1083, Université de Marseille, CINAM UMR 7325 – 2023

10x10e3 spores were incubated for 30 min with 500nM GlowMito in YPD medium.
Credits: Géraldine Leman (Infections Respiratoires Fongiques IRF), & Rodolphe Perrot (Service Commun d’Imageries et d’Analyses Microscopiques SCIAM) – Université d’Angers, France – 2024
")

General enquiries
GlowMito was derived from a triamino-phenazinium belonging to the induline family. With their attractive absorption properties and excellent stability, these molecules have a long history as dyestuffs and attracted major attention in applications from photoreceptor or electrophotography agents to ingredient in color industry.
These derivatives also showed great potential to serve as new mitochondrial probes, although biological applications had so far been neglected. In particular, their appealing photophysical properties characterized by high fluorescence quantum yield and absorption cross-section make them ideal candidates for two-photon excitation microscopy. After having modified some of their substituents to make them internalized by cells and mitochondria more efficiently, GlowMito was born!
The newly synthesized compound, soluble in water, proved very efficient at quickly accumulating within mitochondria without inducing any cytotoxic effects or altering normal mitochondrial functions. Since then, GlowMito expanded the scope of mitochondrial probes by providing a solution of choice to label the entire mitochondrial network quickly and safely in live cells, deep tissues, and even anaerobic organisms.
GlowMito is a cell-permeant probe that is rapidly internalized by cells and specifically target the mitochondria. The quick GlowMito penetration in cells was made possible by the introduction of lipophilic substituents in the original molecule, coupled with its cationic charge, thereby enhancing passive diffusion across cell membranes as it is observed for various lipophilic cations.
As known for various other lipophilic cations, GlowMito accumuation within mitochondria is at least partly driven by membrane potential. Yet, although the precise mechanisms are not yet unraveled, it is likely that GlowMito also relies on other membrane potential-independent mechanisms such as specific interactions with anionic species within the mitochondrial matrix, enhancing the retention of the dye within the mitochondria even with reduced potential.
The strong ability of GlowMito to specifically target mitochondria has been meticulously checked by co-localization studies. More generally, the ability of lipophilic cations to specifically target mitochondria is already well-established and conjugating molecules to lipophilic cations is a commonly used method to develop mitochondria-targeted compounds.
Contrary to classical potential-dependent probes, GlowMito is minimally affected by membrane potential loss and will still produce a visible signal in mitochondria with poor membrane potential. This was demonstrated by pilot studies, later confirmed by early users, showing that GlowMito signal was higher than that of TMRM in presence of an uncoupler. One of our early users even found that GlowMito was able to label hydrogenosomes in the anaerobic organism Trichomonas vaginalis, that have only little -if any- membrane potential, and for which other probes like JC-1, DioC6 and TMRE were not working.
GlowMito produces a bright, red labeling of the entire mitochondrial network, including those with reduced potential. It is perfectly suitable for:
· Live imaging: study of mitochondrial dynamics (velocity, localization), structural changes, multiplexing (potentiometric dyes, calcium signaling probes, etc)
· Downstream analysis: flow cytometry, oxygraphy, etc
We do not recommend its use to measure mitochondrial mass or volume density.
No, GlowMito can be used to label mitochondria in live samples only and is not retained after aldehyde fixation.
So far, GlowMito has been successfully used to label mitochondria in the following biological samples:
· Human cells: HEK293, HeLa, MCF-7, MDA-MB-231, HMLE, UACC-62, U2-OS, Gli36, HAEC, SH-SY5Y, A-172, A549, patient-derived skeletal muscle cells, primary smooth muscle cells & iPSC-derived cardiomyocytes
· Monkey cells: COS-7
· Mice cells: primary cortical neurons
· Tissues: hiPSC-derived heart tissues & mice isolated pressurized blood vessels
· Parasitic protists: Trichomonas Vaginalis
GlowMito showed no internalization in yeast cells
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
Protocol of use
GlowMito comes as a 1 mM stock solution in water, that you can directly dilute in your culture medium. All you need to have on your side is a culture vessel, pre-heated culture medium or your choice and your biological sample.
The full excitation (solid line) and emission (dotted line) spectra of GlowMito in one-photon microscopy is presented below (Ex:542nm/Em:690nm):
For use in flow cytometry, we recommend using a 488nm laser and a 655-730 nm emission filter.
The absorption spectrum of GlowMito in two-photon microscopy is presented below (red line, cross-section = 224GM at 790nm):
The total procedure to label mitochondria in live cells should take no more than 40 min. For deep tissues, total labeling time might need to be extended up to 1.5 h.
The GlowMito signal has been shown to remain stable & specific for up to 3 days with no sign of cell toxicity.
The 1 mM GlowMito stock solution can be stored at 4°C for one month. For long-term storage, aliquot to avoid repeated freeze/thaw cycles and store at -20°C for up to 6 months. Always store GlowMito protected from light.
One vial contains 125µL of a 2,000X aqueous solution for a final volume of 250mL of imaging medium when used at the recommended working concentration. The optimal GlowMito working concentration might need to be increased up to 2-fold when working with deep tissues.
Troubleshooting
GlowMito can be prone to photobleaching when exposed to high-intensity and/or prolonged illumination in one-photon confocal microscopy. Keep in mind that GlowMito photobleaching should be minimal when imaged in 2-photon microscopy.
Here are a few tips you can implement to reduce GlowMito photobleaching:
· Adjust laser intensity and keep it to a minimum.
· Reduce exposure time and try to increase intervals between acquisitions where possible.
Note: capturing a series of short exposure images at regular intervals, instead of a single long exposure, can be used to assess mitochondrial dynamics over time.
· If not sufficient, using anti-fading agents will reduce photobleaching during prolonged imaging sessions.
Note: the ProLong Live antifade reagent has been successfully used in combination with GlowMito and enabled strong conservation of GlowMito signal after 15 sec of continuous light exposure on a confocal microscope.
Fast, reliable and non-toxic solution for measuring bacterial efflux.
ColorFlux is a fast, reliable and non-toxic solution for assessing bacterial efflux for your research on antibiotic resistance. This coloured fluorescent compound quickly accumulates within bacteria as a function of their efflux pump activity. Simply add ColorFlux to your bacteria and watch them change colour !
ColorFlux staining was shown to reflect the activity of well-characterized efflux pumps from the major facilitator superfamily and ATP-binding cassette families in a variety of Gram+ and Gram- bacteria:
– NorA, MepA, MepB, PatA, PatB (Staphylococcus aureus & Streptococcus pneumoniae)
– BmrA (Bacillus subtilis)
– AcrAB (Haemophilus influenzae)
The list will be updated regularly according to the feedback of users.

Tested and validated on : Staphylococcus aureus, Bacillus subtilis, Streptococcus pneumoniae, Haemophilus influenzae.
Colour: Red
Form: liquid
Detection methods: visual & fluorescence. Also compatible with single-cell approaches (live imaging and flow cytometry)
Peak excitation/emission wavelengths: Ex:530nm/Em:650nm
Storage: at 4°C protected from light for 8 months
1 kit contains 1mL of an aqueous solution with a concentration of 1 mg/mL for a final volume of 1L of screening medium

Left graph shows the fluorescence signals detected for each strain using an Ex=530nm, and right graph shows corresponding fluorescence intensity values obtained for each strain at Em=645nm. WT = Wild Type strains, MepA1B+++ = MepA&B overexpressing strains.
Credits: Mrunal Patil & Jean-Michel Bolla, Aix-Marseille Université – 2023

Top picture shows pellet colour variations according to NorA expression. Bottom figures show the fluorescence signals detected for each strain using an Ex=530nm.
Credits: Mrunal Patil & Jean-Michel Bolla, Aix-Marseille Université – 2023

Left pictures show pellet colour variations according to PatA/PatB expression. The right graph show fluorescence signals detected for each strain using an Ex=530nm.
Credits: Mrunal Patil & Jean-Michel Bolla, Aix-Marseille Université – 2023

Top picture shows pellet colour variations according to BmrA expression. Bottom figures show the fluorescence signals detected for each strain using an Ex=530nm.
Credits: Mrunal Patil & Jean-Michel Bolla, Aix-Marseille Université – 2023

Various strains of H. influenzae with known efflux activity were incubated with 1 µg/mL ColorFlux for 25 minutes and centrifuged.
Credits: Cameron Huhn – University of Mississippi Medical School, 2024

Credits: Mrunal Patil & Jean-Michel Bolla, Aix-Marseille Université – 2023

ColorFlux is a cationic compound that quickly penetrates Gram + bacteria through passive diffusion and is efficiently expelled out of the cell depending on efflux activity.
After incubating bacteria with ColorFlux for 15 minutes, a steady-state level is reached and relative levels of efflux activity can be quickly assessed by looking at cell pellet color. It is also possible to read fluorescent signals for a precise quantification of efflux levels. Alternatively, kinetics of ColorFlux accumulation can be monitored by flow cytometry.
ColorFlux is a substrate for efflux pumps that are well-known as key players in bacterial efflux-mediated resistance. In particular, NorA involved in first-line response in S. aureus, PatA/B involved in fluoroquinolone resistance in S. pneumoniae or BmrA, a well-characterized multidrug ABC transporter in B. subtilis.
In a pilot study conducted at IHMT-NOVA, Lisboa on 101 S. aureus clinical strains from humans and animals, ColorFlux detected efflux-resistant bacteria simply by colorimetric assessment. This study demonstrated that ColorFlux can be used as a quick & reliable tool to detect clinically relevant resistant bacteria.
ColorFlux staining was shown to reflect the activity of well-characterized efflux pumps from the major facilitator superfamily and ATP-binding cassette families in a variety of Gram+ bacteria:
NorA, MepA, MepB, PatA, PatB (Staphylococcus aureus & Streptococcus pneumoniae)
BmrA (Bacillus Subtilis)
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
Contrary to ethidium bromide, ColorFlux is a non-toxic compound that you can safely handle on the bench and dispose of. It is a particularly useful tool for training purposes & on-field applications.
The total procedure to stain bacteria and visualise efflux levels by colorimetric assessment should take no more than 30 minutes.
ColorFlux is primarily recommended for Gram+ bacteria as ColorFlux proved efficient at reflecting relative levels of efflux pump activity in all species tested so far. Indeed, the fast influx/slow efflux properties of Gram+ bacteria allow ColorFlux to be quickly internalized and then expelled as a function of efflux, resulting in different shades of red that are representative of relative levels of efflux activity.
Because Gram- bacteria tend to have a slow influx/fast efflux profile, the concentration of ColorFlux and incubation time might need to be adjusted for each particular bacterial species and efflux pump involved. So far, ColorFlux has been successfully used to visually distinguish relative efflux levels of the AcrAB pump in mutant Haemophilus influenzae bacterial strains using the recommended protocol of use. For other species, such as E.coli, some preliminary tests combining ColorFlux with compounds to reduce efflux pump activity, i.e. CCCP, have shown promising results and might help getting relative efflux level measurements.
To sum up, the applicability of ColorFlux in Gram- bacteria will depend on the intended application. While ColorFlux can be useful as a tool to quickly check for the presence/absence of efflux pumps (i.e. to validate efflux pump knock-out mutants), some protocol optimization will be required if you are aiming at comparing relative efflux activity levels between strains, or to detect resistant bacteria.
DNAbsolute is a precipitating reagent for quick & safe DNA extraction of a wide range of samples
DNAbsolute provides a fast & safe method to isolate DNA from a wide range of sample types. Its high sensitivity makes it a solution of choice when working with small & precious samples.
| Product | Product Size | Catalog No. |
| DNAbsolute – column-free DNA extraction buffer | 5ml | ABS-5-IDY |
| DNAbsolute – column-free DNA extraction buffer | 25ml | ABS-25-IDY |
| DNAbsolute – column-free DNA extraction buffer | 50ml | ABS-50-IDY |
Safe & simple
Based on ionic liquids, the DNAbsolute method does not involve any plastic columns nor harmful chemicals. DNAbsolute is the go-to solution for researchers seeking a rapid, user-friendly, and eco-conscious DNA extraction solution.
Versatile
Since its release, DNAbsolute has been successfully used to extract DNA from a wide panel of samples, including some of the most challenging ones:
Insects (drosophila, psyllids, ants, beetles, butterflies, cochineals)
Fish (shark fins)
Mammals (rodent skin)
Plants
Corals
Bacteria (Gram -)
Mollusks
Cnidarians
Protists
Sensitive
Allowing efficient DNA recovery from single specimens down to a single leg, DNAbsolute is a solution of choice to get the most of museum specimens, limited or precious samples.
| 5mL kit |
|
25 to 250 extractions* 5mL DNAbsolute 3M 5mL PBS 150 mM, pH 7.2 5mL Tris 25 mM, pH 8 12mL Lysis buffer 1 12mL Lysis buffer 2 |
| 25mL kit |
|
125 to 1,250 extractions* 25mL DNAbsolute 3M 25mL PBS 150 mM, pH 7.2 25mL Tris 25 mM, pH 8 2x 25mL Lysis buffer 1 2x 25mL Lysis buffer 2 |
| 50mL kit |
|
250 to 2,500 extractions* 50mL DNAbsolute 3M 2x 25mL PBS 150 mM, pH 7.2 2x 25mL Tris 25 mM, pH 8 2x 50mL Lysis buffer 1 2x 50mL Lysis buffer 2 |
*The number of extractions per kit depends on your starting sample weight. For samples weighting from 50 mg to 100 mg, refer to the lowest number of extractions provided. This number will then be inversely proportional to the weight, and can be multiplied by up to 10 when working with samples under 10 mg.
Storage: PBS, Tris & Lysis buffers are stable for 1 year at room temperature (15-25°C). The 3M DNAbsolute reagent shoud be stored at 4°C and is stable for one year.

DNA pellet obtained from 50 drosophila after DNAbsolute precipitation

DNA was extracted from 50 drosophila using either a column-based commercial kit (left column) or DNAbsolute (middle and right columns). The resulting DNA was used for PCR amplification of mTOR gene and amplified products were run on an electrophoresis gel.

DNA was extracted from 50 drosophila using either a column-based commercial kit (left column) or DNAbsolute (right column). The resulting DNA was run on an electrophoresis gel.

Credits: Gabriel Johnson – Smithsonian Institution, Museum Support Center, Maryland, US

Plant leaves either from herbarium specimens (canellaceae – canellaceae & pleodendron) or dried using silica gel (Nyctaginaceae – Pisonia) were extracted using DNAbsolute and run on an agarose gel. DNA yields were quantified on a Qubit instrument.
Credits: Gabriel Johnson – Smithsonian Institution, Museum Support Center, Maryland, US

DNA from ethanol-preserved (1-6) or dried (7-8) Corallium rubrum specimens was extracted using DNAbsolute and diluted 1:10 before serving for PCR amplification of a genomic portion.
Credits: Eugénie Kreckelbergh – Université d’Aix-Marseille, MIO (Institut Méditerranéen d’Océanographie) et FAO (Food and Agriculture Organization of the United Nations).

DNAbsolute is composed of a ionic liquid that has a selective and high affinity for DNA strands. When mixed with your sample, the DNAbsolute reagent will directly bind DNA and precipitate it without the need for RNase treatment, protein precipitation or use of hazardous reagents.
DNAbsolute has been successfully used for extracting DNA from:
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
Yes, DNAbsolute is efficient to isolate DNA from dried samples. So far, DNAbsolute has shown efficient DNA extraction from dried corals and plants (either herbarium specimens, or fresh material dried using a freeze-dry method or silica gel).
Yes, DNAbsolute is efficient at recovering HMW DNA fragments ( >48kb for dried corals, >24kb for arthropods).
DNAbsolute DNA extraction method can yield highly pure DNA due to its ability to efficiently and specifically solubilize DNA, minimizing contamination with proteins, RNA, and other cellular debris. The purity of drosophila and coral DNA extracted using DNAbsolute has been assessed by spectroscopic measurements. The average 260/280 ratio was found between 1.8-1.9 and the 260/230 ratio at 2.3.
The type of biological sample being processed can affect the extracted DNA purity. Sample lysis optimization, or additional purification steps, might be implemented when working with samples containing high levels of secondary metabolites.
A white pellet visible to the naked eye should form immediately after adding the DNAbsolute reagent to your sample lysate. This way, you can easily and rapidly check if your DNA successfully precipitated before proceeding to purification and washing steps.
As a general rule, DNAbsolute is efficient at extracting DNA from a starting solution with a concentration of at least 5 ng/mL. DNAbsolute proved successful at extracting DNA from small starting material, including individual insect specimens (cochineals, psyllids & ants), a couple of legs (butterfly) and even down to a single leg (beetles). As an example, DNA yields from fresh individual cochineals were between 5-155 ng/µL, and between 5-400 ng/µL for a group of 6 individuals.
Moreover, the high specificity of DNAbsolute reagent for DNA means you can safely get pure DNA without additional steps of RNase treatment or protein precipitation, so DNAbsolute is an interesting option if your starting material is not the cleanest.
DNAbsolute is efficient at recovering DNA fragments >300 bp, and will therefore work best with fresh material or material that has been stored immediately at -20°C or below with limited thawing & freezing cycles.
Once your sample is lysed, the whole DNA extraction process should take no more than 30 minutes.
Our DNAbsolute kit contains the DNAbsolute solution as well as some lysis, PBS and Tris buffers. All you need to have on your side is a refrigerated centrifuge, ethanol and proteinase K. You can also add an RNase treatment if needed.
The DNAbsolute reagent is used to purify DNA from a whole lysate. The lysis buffer provided in the DNAbsolute kit is suitable for lysing a wide range of sample types (Gram – bacteria, soil, whole organisms, etc) prior to DNA purification with DNAbsolute. Alternatively, there is no problem in using your own buffer and method of choice for lysis. You can then add the DNAbsolute reagent to your lysate to purify the DNA.
DNAbsolute is available as a 5 mL, 25 mL or 50 mL solution allowing respectively up to 250, 1,250 or 2,500 extractions. Those numbers have been calculated based on 50 drosophila per extraction, and need to be adjusted depending on the quantity and volumes of your starting material while respecting the proportionality of the buffers used.
Yes, DNAbsolute has been successfully used to extract plant DNA from dry leaves (either herbarium specimens, leaves dried using a silica gel or freeze-dried). The method works very well with low starting material (down to 1.5 mg, or a single disk leaf), and the obtained DNA has been successfully used for PCR applications. It is useful to keep in mind that the obtained DNA quality will be very much dependent on your starting material quality, preparation & lysis methods. If particularly high quality standards are needed for your downstream application (i.e. sequencing) it could be that some protocol adjustments are needed (for example, additional subsequent purification steps).
Since its initial launch, DNAbsolute has been heavily used for marine environmental biology. Indeed, DNA extracted from coral specimens (both ethanol-preserved or dried), mussels or even cnidarians. The extracted DNA was directly used for both PCR and qPCR assays after a 1:10 dilution.
The resulting yield can vary widely depending on the sample type and preparation. As a general rule, you can expect to recover on average 85% of DNA from a solution ranging from 10 to 100 μg/mL of DNA, and 60% of a solution ranging from 5 to 10 ng/mL of DNA. Poor quality, and/or fragmented starting material will result in reduced yield of purified DNA.
Here are some yields obtained for different types of samples:
The DNAbsolute ionic liquid has been specifically optimized for double-stranded DNA recovery. We do not recommend using it for RNA isolation.
Kit contents:
– 1x Depigmentation Mix
– 1x Clearing Base
– 1x Imaging Solution
UbiClear is an innovative method for optical clearing of biological samples. Thanks to its unique formulation, the UbiClear solution makes thick samples transparent in less than 20 minutes without any toxic compounds. Simply place your sample in the clearing buffer, incubate at 37°C and transfer it to the imaging solution. A great way to make clearing accessible to anyone!
Ready to dive into deep tissue imaging ?
Non-toxic
Formulated without any harsh chemicals, UbiClear can be handled safely and does not deteriorate microscope objectives
Compatible with fluorescence labeling
UbiClear preserves endogenous fluorescence and can be combined with immunostaining protocols
Ultra-fast
Stop spending days on clearing: clear your samples within minutes thanks to a 1-step clearing method
Versatile
Clears a vast panel of soft & hard mammalian tissues (bone, teeth), insects & plants. Nothing can stop it!
UbiClear is an aqueous solution that modifies the optical properties of complex biological tissues by removing lipids and homogenizing refractive indexes. This renders opaque tissues transparent while keeping their original structure intact. As a result, light passes through specimens without any scattering or absorption, yielding high-quality images of entire 3D structures. The UbiClear workflow is straight-forward, and does not require any specific expertise or access to sophisticated equipment.
Step 1
Remove sample coloration by immersing it into UbiClear Depigmentation Buffer for 1h at 37°C.
Step 2
Clearing (20min – overnight)
Incubate your sample into the UbiClear clearing solution at 37°C until fully transparent.
Maximal transparency is achieved in a few minutes for most tissue fragments (<20 min for mouse cerebral cortex and small intestine), and may take up to a few hours for entire organisms.
Step 3
Imaging
Transfer your cleared sample to the imaging solution (RI = 1.47) for imaging and maintaining transparency on the long-term.
Step 4
Repeat !
If needed, samples can be re-opacified simply by transferring them to 0.2 M Tris buffer.
Discover more on Ubiclear & its applications by hearing directly from its inventors: http://brg.sppin.fr
Sample types: UbiClear can be used to clear a wide spectrum of samples ranging from small 3D cell structures up to entire organisms. So far, UbiClear has shown efficient clearing of:
– 3D cell structures: human embryoid bodies, human mini-brains
– Human tissues: frontal cortex
– Mice tissue sections (up to 1 mm thick): spinal cord, brain (cerebellum, choroid plexus), testes, seminal vesicle, ear (elastic cartilage and hair), lungs, heart, intestine, kidney, skeletal muscle, yolk sac membranes
– Ox tissues (up to 1 mm thick): knee (joint cartilage and bone)*
– Plants: ivy leaves, carrot slices, beet roots, microcallus & callus organoids, nasturtium stamen, melon seeds, sprouted tomato seeds, rose stem slices
– Whole organs: mouse brain, heart, intestine, eye, teeth*, dorsal root ganglia
– Whole organisms: Colisa Lalia, adult zebrafish, firebugs (Pyrrhocoris apterus), wasps, grasshoppers, shield bugs (Graphosoma lineatum), flies, cockroaches, spiders
– Crustaceans: woodlouse
– Molluscs: mussel (including shell)*
*for these samples, an additional step of demineralization needs to be performed before clearing.
Compatible assays: label-free anatomical studies (autofluorescence, volumetric imaging), dye-based assays (Evans blue, albumin-FITC), immunostaining. UbiClear preserves endogenous fluorescence.
Microscopy techniques: confocal, spinning-disk, light-sheet microscopes*.
*Extra diluting buffer can be purchased separately to make more imaging solution if needed (i.e. for use with light sheet microscopes).

Storage: The depigmentation buffer, Mix 1 and Mix 2 solutions should be kept at room temperature, protected from light. In these conditions, all solutions can be stored for at least 1 year.
| Small kit |
| Depigmentation Buffer: 20 mL Mix 1: 5 mL Mix 2: 40 mL |
| Final volume of depigmentation, clearing & imaging solutions = 20 mL each* |
| Large kit |
| Depigmentation Buffer: 200 mL Mix 1: 50 mL Mix 2: 400 mL |
| Final volume of depigmentation, clearing & imaging solutions = 200 mL each* |
*Mix 1 & Mix 2 are mixed at a 1:5 ratio to make the final clearing solution. Mix 2 will be added with water to make up the final imaging solution.
An extra 400 mL volume of Mix 2 can be purchased separately to make more imaging solution if needed (i.e. to fill out light sheet microscope tanks). A final volume of 400-500 mL of imaging solution can be prepared using the 400 mL of Mix 2 provided.
The volume of clearing solution required varies according to your sample size. As a general rule, we recommend using a clearing solution volume corresponding to 10X your sample volume. Please refer to the table below to find out which kit format will best suit your needs.


1 mm thick mice brain slices (fat tissue), 2 cm mice intestine fragments and a 1 mm thick ox knee slices (joint cartilage and bone, calcified tissue) were cleared using the UbiClear method and imaged on a striped target. Ox knee joint samples were decalcified and all samples depigmented and cleared using UbiClear. The target stripes are visible through the sample after clearing for less than an hour.
Credits: Brigitte Delhomme & Martin Oheim (CNRS, SPPIN UMR8003)
A 1.5 cm long mice small intestine fragment was cleared using UbiClear and deep autofluorescence images were taken using a two-photon microscope at different depths of the intestine wall, starting from the muscularis longitudinalis down to the crypts. The video shows the 3D rendering and animation of the fine architecture of the enteric nervous system based on a z-stack acquired (800 nm excitation, z-step: 1 µm, 480 ms/image).
Source publication: Hazart D. et al, 2023

Indirect immunofluorescence and clearing was performed on 1 mm thick sections of formaldehyde-fixed mouse brain using GFAP antibody and secondary antibody Alexa 555 fluorophore. On the left the whole slice was imaged in mosaic with 10XNa0.3 objective, on the right 40XNa1.0 was imaged in the cortex of the mouse brain.
Credits: Diane Derrien, Brigitte Delhomme & Martin Oheim (CNRS, SPPIN UMR8003)

Immunofluorescence and clearing was performed on whole fixed dorsal root ganglia from a transgenic mouse model that expresses both (i) a genetically-encoded GCaMP6 calcium sensor in sensory neurons as well as (ii) a hematoglutinin-tagged chemogenetic receptor in satellite glial cells. An anti- GFP (green) and an anti-hematoglutinin (red) antibodies were used to amplify GCaMP6 staining and reveal satellite glial cells, respectively. The whole ganglia was imaged using a confocal microscope Axio LSM710 (Zeiss) and 10XNa0.3 WD 5mm air objective, in 620 µm depth and z 2 µm. 3D reconstruction was obtained with Imaris viewver software (oxford instruments).
Credits: Andreia Ramos (Université Paris Cité, CNRS, INCC UMR8002, Cendra Agulhon’s Group) and , Brigitte .Delhomme (Université Paris Cité, CNRS, SPPIN UMR8003, Martin Oheim’s laboratory). Courtesy of Cendra Agulhon.

Immunofluorescence was performed on a 500µm mouse spinal cord section after depigmentation. Iba1 antibody, a rabbit polyclonal antibody, and Cy3 secondary antibody were used for incubations. Iba1 was imaged using an Axio LSM710 (Zeiss) confocal microscope with 10XNa0.3 WD 5mm air objective, zoom 0.6 in 270µm depth and z 2µm. 3D reconstruction was performed using Imaris viewver software (Oxford Instruments).
Credit : Clearing B.Delhomme & M.Oheim (CNRS, SPPIN UMR8003)

A firebug (Pyrrhocoris apterus) was depigmented, embedded in low melting agarose and cleared using UbiClear. The image showing autofluorescence signal at 488 nm was acquired using a light-sheet ultramicroscope (Labvision). 2,400 images were stitched for the 3D reconstruction.
Credits: Clearing: Brigitte Delhomme (CNRS, SPPIN UMR 8003), Imaging: J. Fernandes (Institut Pasteur, Paris) & Martin Oheim (CNRS, SPPIN UMR8003)

A whole brain from a 4-month old mouse was depigmented and cleared using UbiClear. The picture was taken 48h after clearing.
Credits: Brigitte Delhomme & Marwa Moulzir (CNRS, SPPIN UMR 8003)

A 1.5 cm long mice intestinal fragment was cleared using UbiClear and placed in a home-made tank with a black-and-white stripe pattern. Images were acquired on a SMZ800 stereo macroscope (Nikon, Amstelveen, The Netherlands) after 10sec, 110sec and 20min of incubation in the UbiClear clearing solution. Michelson’s contrast calculation revealed that 71% transparency was reached in less than 15 min.
Source publication: Hazart D. et al, 2023

Mouse was perfused with FITC albumin and sacrificed to image blood vessels in the cerebellum. A 500µm tissue slice was cleared without depigmentation and imaged using an Axio LSM710 (Zeiss) confocal microscope with 488nm laser and 10XNa0.3 WD 5mm air objective, in 508µm depth and z 4µm. 3D reconstruction was performed using Imaris viewer software (Oxford Instruments).
Credits : Mouse painting vessels X.Bai (University of Saarland, Homburg (DE)), Clearing B.Delhomme (CNRS, SPPIN UMR8003, Paris (Fr)) , F.Licata (UMS Paris-cité University (Fr) & Martin Oheim (CNRS, SPPIN UMR8003, Paris (Fr))

Mice small intestine fragments were cleared using UbiClear and immunolabeled against a pan-neuronal marker (red) and an enteric-glia marker (green). Single autofluorescent images (grayscale) and fluorescence images were taken at different depths in the intestinal wall (muscularis longitudinalis, Auerbach’s plexus, Meissner’s plexus and crypts). Images were acquired on a LSM880 inverted confocal Airyscan microscope (Zeiss).
Source publication: Hazart D. et al, 2023
Hazart D, Delhomme B, Oheim M and Ricard C (2023) Label-free, fast, 2-photon volume imaging of the organization of neurons and glia in the enteric nervous system. Front. Neuroanat. 16:1070062. doi: 10.3389/fnana.2022.1070062.
Doriane Hazart, Malvyne Rolli-Derkinderen, Brigitte Delhomme, Pascal Derkinderen, Martin Oheim and Clément Ricard (2024) L’intestin, lanceur d’alerte, dans les prémices de la maladie de Parkinson. Med Sci (Paris), 40 6-7 (2024) 544-549. doi: https://doi.org/10.1051/medsci/2024082
Discover all posters featuring UbiClear at the AFH (French Association of Histotechnology) congress:
UbiClear relies on an aqueous clearing solution, formulated without any harmful solvents or expensive components, allowing to make thick biological samples transparent in no time. By providing a safe and affordable technique even when large volumes are needed, UbiClear was designed to democratize tissue clearing for researchers interested in deep tissue imaging.
UbiClear can be used to clear a wide spectrum of samples ranging from small 3D cell structures up to entire organisms. So far, UbiClear has shown efficient clearing of:
– 3D cell structures: human embryoid bodies, human mini-brains.
– Human tissues: frontal cortex.
– Mice tissue sections (up to 1mm thick): spinal cord, brain (cerebellum, choroid plexus), testes, seminal vesicle, ear (elastic cartilage and hair), lungs, heart, intestine, kidney, skeletal muscle, yolk sac membranes.
– Ox tissues: knee (joint cartilage and bone)*.
– Fish tissues: Colisa Lalia, zebrafish.
– Plants: ivy leaves, carrot slices, beet roots, microcallus & callus organoids, nasturtium stamen, melon seeds.
– Whole organs: mouse brain, heart, intestine, eye, teeth* and dorsal root ganglia.
– Insects: firebugs (Pyrrhocoris apterus), wasps, grasshoppers, spiders, shield bugs (Graphosoma lineatum), flies, cockroaches.
– Crustaceans: woodlouse.
*for bone & teeth samples, an additional step of demineralization needs to be performed prior clearing.
We would greatly appreciate your valuable inputs by helping us complementing this list with any of your positive research experiences. Drop us a message through our Contact page!
Yes, it is possible to re-opacify the samples by incubating them in a 0.2 M Tris buffer at pH 8. Re-opacifying can be a practical way to select and section a region of interest for downstream assays (i.e. immunostaining).
The final imaging solution obtained following our protocol of use (i.e. diluting the Mix 2 solution in water) has a refractive index of 1.47. It is possible to increase it up to 1.485 by adding less water to the Mix 2 solution.
UbiClear efficiently clears biological tissues by removing lipids (Mix 1) and homogenizing refractive indexes (Mix 2) of the sample’s internal components. The clearing solution is prepared by mixing Mix 1 & Mix 2, and the imaging solution is prepared by diluting Mix 2 in water.
Yes, endogenous fluorescence is preserved upon clearing (this has been shown with dT-tomato and GFP). If your sample contains a fluorescent native protein, you simply need to skip the optional depigmentation step as this will likely interfere with the fluorescence signal.
Yes, it is possible to perform immunostaining and clear your labeled sample with UbiClear afterwards.
Immunostaining using AF405, AF488, AF555, AF647, Cy3, TMR, AM-cyan &Td tomato have been successfully performed on samples cleared with UbiClear.
Yes, UbiClear can be combined with direct dyes for visualizing cellular or structural features. This has been validated with Evans blue and albumin-FITC for imaging blood vessels in cleared samples. UbiClear is not compatible with common direct nucleic acid dyes (DAPI, BET, methyl green, Topro3…). If DNA staining is expected, we recommend using indirect antibody labeling strategies.
So far, the UbiClear kit has been successfully used on samples cleared using 10% formalin (or 4% PFA), and that is therefore the fixation method we recommend. It could be that clearing works well with other fixation solutions, yet we can’t guarantee that clearing efficiency will be similar.
Once cleared, samples can be imaged using conventional confocal microscopes (for samples up to ~500 µm thick). Thicker samples may be imaged using light-sheet microscopes.
Need some advice? We would be happy to answer any questions on the choice of microscopy technique, data analysis and handling. Feel free to reach out at technical@stratech.co.uk
Total duration will vary depending on your sample thickness and internal properties. As a general rule, the total procedure should not take more than 2 hours for samples up to 1 mm thick. The protocol might be extended up to a few hours for entire organisms.
As UbiClear works by removing lipids from the samples, this solution will not be suitable for imaging & staining lipids. It is also not compatible with direct nucleic acid dyes (DAPI, BET, methyl green & Topro3). If DNA staining is expected, we recommend using indirect antibody labeling strategies.
Sample shrinkage has been observed within the first few minutes of incubation in the clearing solution, which is explained by the difference in osmolarity between the sample and the solution leading to dehydration. This effect is then attenuated after a few minutes as the clearing solution penetrates inside the structures.
No, UbiClear does not contain any organic solvents neither volatile compounds. Its handling does not require the use of a chemical hood or sophisticated ventilation systems for imaging.
The UbiClear kits include a Depigmentation Mix, a Clearing Base and some Imaging Solution. Everything you need to have on your end is some water, 0.2M Tris at pH 8 and some 30% hydrogen peroxyde.
The volume of clearing solution required varies according to your sample size. As a general rule, we recommend using a clearing solution volume corresponding to 10X your sample volume. Please refer to the table below to find out which kit format will best suit your needs.

Yes, our imaging solution does contains an anti-fading agent used to preserve fluorescence of tissues and cell structures. The Ubiclear imaging solution has been notably used for confocal, light-sheet and 2-photon imaging without any noticeable fading effects. A potential risk of fluorescence decay may arise if the solution is used without adding any water to it (although we recommend adding water at a 1:5 ratio to achieve a refractive index of 1.47, the solution may be used as it is when a higher refractive index is needed).
Additional Mix 2 is usually needed when using microscopes that include a large tank or chamber to be filled with imaging solution (this is typically the case with light-sheet microscopes, or more generally setups where whole organisms or large samples need to be imaged over extended periods). If this is your case, we would recommend buying an additional bottle of Mix 2 to make your required amount of imaging solution. If not, the final volume of imaging solution needed will be similar to that of the clearing solution, so the amount of Mix 2 included in the kit is sufficient.
We recommend using the imaging solution provided in the kit (Mix 2) to ensure that the sample is imaged in a solution of a similar composition to that of the clearing solution used beforehand, and therefore minimize the potential artifacts linked to imaging solution heterogeneity. Indeed, the imaging solution prepared as per the Ubiclear protocol will have the exact same composition as the clearing solution without the detergents to preserve the sample integrity and microscope objectives. Moreover, the Ubiclear solution has been specifically formulated to reduce photobleaching, as well as to re-equilibrate refraction indexes and reverse any potential shrinking that might have happened upon clearing.
Especially, we do not recommend using oils as imaging solutions due to the high water content and poor fat solubility of the clearing solution.
Kit contents:
– Ready-to-use coverslip(s) with your choice of structure design(s)
– Stencell(s) (1 per coverslip) for optimizing substrate coating & multiplying experimental conditions
)")
– Introducing the next generation of nanostructured substrates for cellular applications –
FakirSlide are ready-to-use coverslips patterned with micro- and nano-structures to induce diverse, well-controlled topological cues on live cells or planar model membranes.
Made of borosilicate glass, they are compatible with all types of advanced microscopies and provide a solid support allowing for long-term experiments.
Unlock new insights! Based on a newly-developed nanostructuration technique called soft nanoimprint lithography, FakirSlide pushes the resolution boundaries and makes available to researchers a diverse panel of reproducible micro to nano-scale topographies of various aspect ratios to better investigate key cellular responses to specific membrane topologies. Check out our available catalog below.
Maximize your assay reliability – FakirSlide provides ordered arrays of structures with high control over their shape, diameter and periodicity. They were specifically designed to induce robust & consistent membrane curvatures and facilitate data analysis.
Simplify your experimental workflows: made of a high-quality synthetic silica layer, the Fakirslide surface is directly functional for supported lipid bilayers without the need for harsh cleaning or hydroxylating pre-treatments.
Fakirslides substrates have been successfully used to manipulate membrane morphology of living cells and supported lipid bilayers, and observe effects of curvatures on membrane protein dynamics, cytoskeletal reorganization and cell migration. Check out relevant publications and example results in our dedicated sections.

")
")
")
")
We are thrilled to introduce our newly-released design, the Nanocones, that just joined the FakirSlide catalog :
")
Need help to decide? Check out some example results obtained with our different designs and more tips on how to select your structures in our Results, Publications & FAQ sections.
The FakirSlide technology paves the way to a new kind of nanostructures for biological applications, allowing for high flexibility in designs. It is thanks to your interest and feedback that we will be able to expand this catalog and offer new shapes in the future. Nanoridges, hexagones… Stay tuned !
Use FakirSlide to apply membrane deformations on cultured cells or membrane-mimetic systems. Experimental outputs include:
Live cell imaging
Immunostaining
Migration assays
Compatible imaging modes: confocal microscopy, Airyscan microscopy, TIRF, super-resolution microscopy (2D and 3D STED, PALM/STORM), Scanning Electron Microscopy (SEM), Atomic Force Microscopy (AFM).
Cell types: So far, FakirSlide has been successfully used with a variety of human (HeLa, U-2 OS, HT1080, SUM159, RPE-1, THP-1 & human monocyte-derived dendritic cells (moDC)) and murine (C2C12 mouse myoblasts) cells.
Supported membranes: So far, FakirSlide has been successfully used with a variety of neutral (DOPC, Egg-PC, POPC, POPE & Egg-PE) and negatively-charged (Liver-PI, Brain-PS, Brain-PI(4,5)P2 & Brain-PI4P) lipid mixtures.
Surface topographies: round or square pillars, nanodomes or nanocones
Approx. pattern area: 1 cm x 1 cm
Surface material: borosilicate glass
Coverslip diameter: 25 mm
Coverslip thickness N°: 1.5H (0.170 mm ± 0.005 mm)
Cell culture treated: No
Storage: can be stored indefinitely at room temperature when protected from dust & humidity
Ready-to-use FakirSlide structured coverslips with your choice of nanostructure(s)
Stencells (1 per coverslip) for optimized coating & multiplied experimental conditions, with your choice of design(s)
1. Select your favorite structures
Choose your coverslip(s) among our available catalog, according to your preferred design(s).

")
2. Add some Stencells to your order at no extra cost
Stencells are silicon chambers that you can easily stick and remove from the FakirSlide coverslips to create controlled culture areas. They will help you make the most of your FakirSlide coverslips.
Optimize your coating: get a homogeneous coating while saving reagents by using the Solo, Quartet or Nonet Stencells.
Increase your outputs: don’t need a large surface? Use the Quartet & Nonet designs to multiply your experimental conditions on a single FakirSlide coverslip and create structure-free control areas.
Find out more about Stencells on our dedicated product page.
")
Snapshots of cytoskeleton (spy555-actin, orange) and plasma membrane dynamics (WGA-AlexaFluor488, green) of moDC seeded on round pillars (Large). Images were acquired on a spinning disk microscope (Nikon Ti Inscoper CSU-X1, objective 100X Plan Apo lambda 1.45 NA DT 0.13mm oil).
Image credits: Raissa Rathar – IRIM Montpellier, 2022
")
Confocal and STED images showing the organization of the actin cytoskeleton (STAR 580 phalloidin, Abberior) of moDC cells seeded on round pillars (Large). Images were acquired on an Abberior STED super-resolution microscope (Objective 100X Plan SuperApochromat N.A 1.4 Oil).
Image credits: Raissa Rathar – IRIM Montpellier, 2022

Airyscan sub-diffraction microscopy images of a nano-domes surface (Small) functionalized with supported-lipid bilayers (doped with 0.2% of OG-DHPE, green) along with the organization of the recombinant protein FCHo2 labeled with AlexaFluor647 (protein concentration was 1 µM, magenta). Images were acquired on a confocal/airyscan Zeiss LSM880 microscope (objective 63X Plan Apo Oil 1.4NA).
Image credits: El Alaoui et al., 2022

Airyscan sub-diffraction microscopy images of moDC cells seeded on round pillars (Large) and immunostained to visualize the endogenous organization of paxillin (anti-paxillin rabbit polyclonal antibody GTX125891, orange) and the actin cytoskeleton (phalloidin Atto647N, blue). Images were acquired on a confocal/airyscan Zeiss LSM880 microscope (objective 63X Plan Apo Oil 1.4NA).
Image credits: Raissa Rathar – IRIM Montpellier, 2021

Snapshots of the dynamics of moDCs cells seeded on round pillars (Large). Images were acquired on an inversed Olympus IX83 video-microscope (transmitted light, objective 40 X objective 40x LUCPLFLN 0,6NA RC2) 0.6 NA. Blue arrows point to membrane projections propelled during the migration of moDC cells.
Image credits: Raissa Rathar – IRIM Montpellier, 2021

Left: Average intensity (AVG projection of n > 5 images) of the surface distribution of fluorescent PI(4,5)P2 (gray) and the surface distribution of 100 nM wild-type, ΔPB-Cdc11 or ΔAH-Cdc12 septin-GFP complexes (Fire LUT) at the top and bottom of cones functionalized with 2.5% mol PI(4,5)P2-containing membranes. Scale bar, 1 µm. Right: Enrichment of wild-type, ΔPB-Cdc11 or ΔAH-Cdc12 mutants septin-GFP complexes at the top (k ~6.6µm-1), middle (k ~2 µm-1) and bottom (k ~1.4 µm-1) of nano-cones relative to the flat membrane (k ~0µm-1). Nano-cones analyzed, n = 265, 277, 342, and 580; n = 269, 279, 376 and 540; n = 266, 275, 315 and 586, for wild-type, ΔPB-Cdc11 or ΔAH-Cdc12 mutants respectively, from three technical replicates. Error bars represent s.d.; One–way ANOVA test: n.s > 0.05, *P < 0.05, ** P < 0.01, **** P < 0.0001.
Source publication: El Alaoui et al, Septin filament assembly assist the lateral organization of membranes, bioRxiv 2024; https://doi.org/10.1101/2024.03.19.585775

Immature hDCs were cultured on Fakirslides (round pillars). a. Scanning electron microscopy images showing a detail of the surface topography (left) and the deformation of the plasma membrane of hDCs to adapt the shape of the surface topography (right). Scale bar: 1.5 and 3 μm, respectively. Schematics generated by BioRender. b. Representative Airyscan images showing the endogenous organization of the receptors (orange hot LUT) CD64, Dectin 1, DC-SIGN, and TLR4 relative to podosomes (cyan hot LUT) at the plasma membrane of immature hDCs on flat or vertical pillar topographies. Scale bar, 2 μm. c. Interaction strength (ε) of CD64, Dectin 1, DC-SIGN, or TLR4 with the actin signal at the plasma membrane of immature hDCs seeded on flat (gray) or vertical pillar topographies (red). Solid line represents the mean. Cells analyzed (n) from 2 biological replicates, n = 16, n = 15, n = 17, n = 13 for flat and n = 13, n = 19, n = 12, n = 15 for pillar topographies, respectively. Mann–Whitney test: n.s. P > 0.05, **P < 0.01.
Source publication: Rathar R. et al, (2024). Tuning the Immune Cell Response through Surface Nanotopography Engineering. Small Science. 10.1002/smsc.202400227.
2020 | Micro/Nanostructure Engineering of Epitaxial Piezoelectric α-Quartz Thin Films on Silicon
Qianzhe Zhang, David Sánchez-Fuentes, Rudy Desgarceaux, Pau Escofet-Majoral, Judith Oró-soler, Jaume Gázquez, Guilhem Larrieu, Benoit Charlot, Andrés Gómez, Martí Gich, and Adrián Carretero-Genevrier. ACS Applied Materials & Interfaces 2020 12 (4), 4732-4740DOI: 10.1021/acsami.9b18555
2020 | Mapping cell membrane organization and dynamics using soft nano-imprint lithography
T. Sansen, D. Sanchez-Fuentes, R. Rathar, A. Colom-Diego, F. El Alaoui, S.de Rossi, J. Viaud, M. Macchione, S. Matile, R. Gaudin, A. Carretero-Genevrier, and L. Picas, ACS Appl. Mater. Interfaces 2020. doi.org/10.1021/acsami.0c05432
2022 | Structure and dynamics of FCHo2 docking on membranes
Fatima El Alaoui, Ignacio Casuso, David Sanchez-Fuentes, Charlotte Arpin-Andre, Raissa Rathar, Volker Baecker, Anna Castro, Thierry Lorca, Julien Viaud, Stephane Vassilopoulos, Adrien Carretero-Genevrier, Laura Picas. eLife 2022;11:e73156 doi: 10.7554/eLife.73156
2024 | Septin filament assembly assist the lateral organization of membranes
Fatima El Alaoui, Isabelle Al-Akiki, Sandy Ibanes, Sébastien Lyonnais, David Sanchez-Fuentes, Rudy Desgarceaux, Chantal Cazevieille, Marie-Pierre Blanchard, Andrea Parmeggiani, Adrian Carretero-Genevrier, Simonetta Piatti and Laura Picas. bioRxiv; https://doi.org/10.1101/2024.03.19.585775
2024 | Tuning the Immune Cell Response through Surface Nanotopography Engineering
Raïssa Rathar, David Sanchez-Fuentes, Hugo Lachuer, Valentin Meire, Aude Boulay, Rudy Desgarceaux, Fabien P. Blanchet, Adrian Carretero-Genevrier, Laura Picas. smallscience; https://doi.org/10.1002/smsc.202400227
This publication has been promoted in this newly-released article on the CNRS website.
")
Learn more about how FakirSlide can benefit your research:
FakirSlide rely on a newly-developed method based on soft nanoimprint lithography on sol-gel thin films. This bottom-up approach allows the reproducible patterning of ordered arrays of nanopillars with different diameters and heights on glass subtrates.
By providing ready-to-use coverslips, FakirSlide opens up the applications of bottom-up lithographic procedures to researchers, that were so far constrained due to the requirement of complicated and limited-access technology.
The use of soft nanoimprint lithography on sol-gel thin films, the production technique used for the Fakirslides, allows the formation of a wide variety of structure sizes and shapes that cannot be achieved using the focused ion beam technique (for example the nano-cones).
Moreover, it allows to create much larger structured surface areas (1 cm2 per coverslip) compared to what is usually achieved using focused ion beam methods.
The structures obtained on the Fakirslides are also perfectly repeatable, from one structure to another but also from one coverslip to another. Structure reproducibility can be challenging with the focused ion beam technique.
Finally, focused ion beam methods often require access to sophisticated platforms, expertise or imply finding collaborations. The Fakirslides were designed to provide a ready-to-use solution, so that you can start your work right-away and spend your time for what really matters.
Made of borosilicate glass, the FakirSlide coverslips are the substrates of choice for super-resolution microscopy applications. They are compatible with advanced microscopy experiments without a risk of compromising the quality of imaging across nanopillar surfaces due to refractive index mismatching.
Using a glass-based substrate also offers more rigid structures (Young’s modulus ~64 GPa), providing increased control over cellular membrane bending and improved overall solidity.
Finally, the application of soft nanoimprint lithography provides access to a wide panel of micro- to nano-structures with various aspect ratios, which fabrication was previously constrained due to the requirement of complicated and limited-access technology.
We have developed a wide panel of micro- and nano-structures for both cellular and isolated membrane applications. Here is a short overview of each design specificities and some use cases.
These patterns have been commonly used to induce controlled membrane deformations in cellulo (i.e. with live cells).
They are particularly useful to study cellular response to specific mechanical cues associated with membrane topologies: cytoskeletal organization, recruitment and/or disassembly of membrane-associated proteins or proteins holding a curvature sensing domain, or cell migration.
Earlier studies using FakirSlide have shown that structural arrangement of proteins at the cell surface is imposed by the morphology and periodicity of the substrates. Therefore, combining several designs in a single kit will allow you to investigate the precise effects of nanostructure size and/or shape on cellular responses. Example of relevant design combinations include:
A “shape” exploratory pack: Square nanopillars + Small round pillars + Small nanodomes
Check out this publication showing isotropic organization of AP-2 protein and clathrin around small round pillars and a remarkable anisotropic arrangement on square pillars.
A “size” exploratory pack: Small round nanopillars + Large round nanopillars
Check out this publication showing how the round pillar size differentially influences the distribution of the surface receptor TfR as well as the recruitment of endocytic F-BAR protein FCHo2.
Alternatively, given the coverslip design, it is possible to include a flat control surface as a non-structured control area in parallel to your nanostructured area. This can be an interesting option to assess the impact of a single nanostructure pattern.
Check out our Results section to discover how nanostructured surfaces support membrane projections during the migration of dendritic cells.
smaller in size, these structures are particularly recommended for inducing curvatures on isolated membranes. In addition, the smooth edges of the nanodomes make them a substrate of choice for AFM microscopy.
Check out this publication showing the use of FakirSlide small nano-domes to study the docking & self-organization of the endocytic F-BAR protein FCHo2 on curved membranes. In this study, small nano-domes were used to reproduce the dome-like invagination that is a major step in the formation of clathrin-coated structures.
Check out this publication showing the use of FakirSlide small nano-cones to investigate the curvature-associated localizations of septin complexes on plasma membranes using reconstituted systems.
The FakirSlide nanostructured substrates have be used to manipulate the shape of cellular membranes and obtain unprecedented outputs including, amongst others:
For more detailed examples of FakirSlide uses, check out example results and full publications in our dedicated sections.
The FakirSlide coverslips are made of borosilicate glass, and are therefore perfectly compatible with any kind of advanced microscopy including confocal microscopy, Airyscan microscopy, TIRF, 2D and 3D STED, PALM/STORM, Scanning Electron Microscopy (SEM) and Atomic Force Microscopy (AFM).
The FakirSlide coverslips can be cleaned using sterile water and UV-light if needed. We do not recommend using aggressive solutions (i.e. piranha, other acids, strong bases or polar solvents) as they may damage the nanostructured silica layer.
Note that the Fakirslides coverslips are made of a synthetic glass using a sol-gel process adn fabricated in a clean-room environment. This results in substrates with a high cleanliness level, which do not necessitate any harsh cleaning (i.e. NaOH, piranha bath, etc) as it can be the case with standard commercial coverslips.
Yes, the FakirSlide coverslips can be coated with the adhesive protein of your choice for cellular applications just like other commercial glass surfaces.
If a hydroxylating treatment is needed to immobilize specific molecules at the surface, we recommend gentle hydroxylation methods to minimize impact on the surface structure (i.e. thermal treatment, plasma treatment or UV/ozone treatment).
Yes, the Fakirslides can be used to make supported lipid bilayers without the need for cleaning or hydroxylating pre-treatments. Indeed, the structured surface layer is made of a synthetic silica material which structuration method involves a pirhana bath treatment and is performed in a clean-room facility. The resulting surface can be used directly for supported lipid bilayer applications, and shows similar performances in terms of lipid bilayer formation compared to pirhana-treated standard commercial coverslips.
If an additional treatment is needed to immobilize specific molecules at the surface, we recommend avoiding aggressive solutions (i.e. piranha, strong acids or polar solvents) to minimize the risk of altering the nanostructures.
Other questions? Please contact us.
")









")




")




")
")
")

)")
")