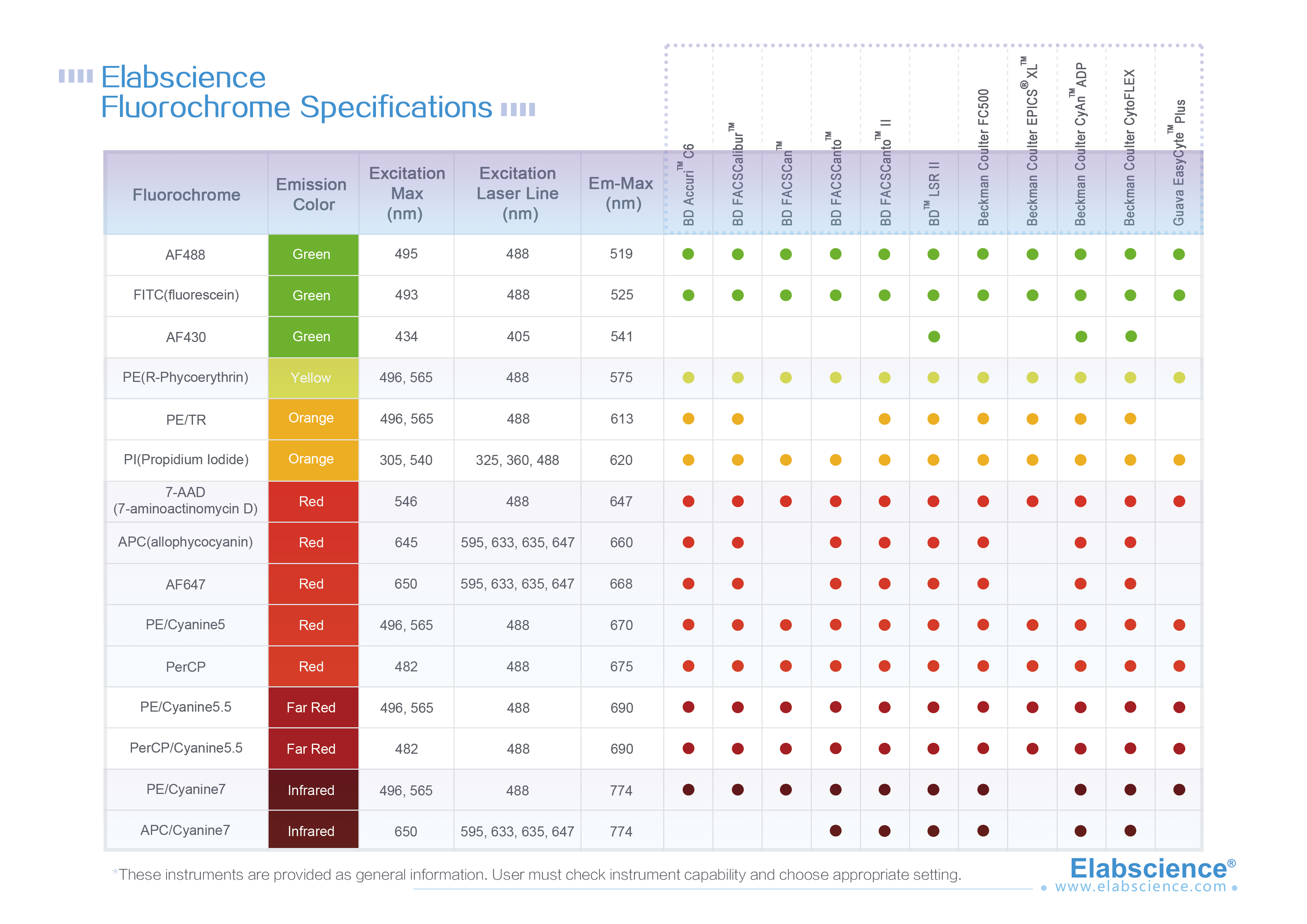
Single cell is a prerequisite for analysis and detection of cells with flow cytometry (FCM). Therefore, solid tissue must be prepared into single cell suspension. In FCM, the preparation of single cells is an important step. It requires that tissue should be dispersed into single cells, but also that the single cells should maintain their inherent biochemical components and biological characteristics.
The basic principles of flow cytometry sample preparation are as follows.
1). Make sure that the cell samples are fresh, and sample preparation and detection should be finished as soon as possible.
2). Appropriate washing, enzyme digestion or EDTA treatment should be applied for different cell samples to remove impurities and detach adhesive cells to form single cell suspension.
3). Enzyme digestion, mechanical dispersion and chemical dispersion can be applied to tumor tissue to obtain single cell suspension.
4). The paraffin embedded tissue should be cut into 40~50 μm thick slices first. After dewaxing to water, the single cell suspension can be prepared with the previous method.
5).The number of single cells in suspension should not be less than 107/mL.
The experimental operation of flow cytometry can be divided into five steps.
1). Sample acquirement. Select typical surgical or biopsy tissue. For example, for the surgical tumor the vigorously growing part should be the first option. The tissue and other specimens should be kept fresh after separation. Usually the samples should be treated within 1 h at room temperature or preserved in fixative or at low temperature in time.
2). Staining cells with fluorescence antibodies stain the cells.
3).Loading samples for detection and data acquirement with software program provided by the manufacturer.
4). Quantitative analysis of the data.
5). Evaluation of the biological and medical significance the results.
1. Preparation of peripheral blood samples
1) General preparation of peripheral blood samples
a. Collect peripheral blood with heparin anticoagulation. Samples should be stored at room temperature (25°C), and treated within 6 h.
b. ACK buffer should be restored to room temperature before use.
c. Add 200μLperipheral blood into the flow tube, then add 3 mL ACK buffer into the tube and incubate for 3~5 min at room temperature.
d. Add 10 mL cell staining buffer (or PBS with 0.1% BSA) to stop lysis.
e. Centrifuge at 300 g for 5 min at room temperature. Discard the supernatant.
f. Repeated the above washing step for another time.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
2) Preparation of mononuclear cells with peripheral blood samples
a. Collect 2 mL peripheral blood with heparin anticoagulation and bring the volume to 4 mL with physiological saline.
b. Add 4 mL (or equal volume of the peripheral blood sample to be treated) lymphocyte separation solution to the 15 mL conical tube.
c. Add equal volume of peripheral blood along the wall of the tube slowly to make sure that the peripheral blood is above the liquid level of the lymphocyte separation solution. Do not add too fast to prevent the blood from mixing with the separating solution.
d. Centrifuge at 400 g for 20~30 min at room temperature (The centrifuge conditions are according to the blood samples, it is suggested to find out the best separation condition before the experiment). After centrifugation, the blood in the tube is clearly divided into 4 layers. The upper layer is the plasma, the second is the white Lymphocytes, the third layer is the transparent separating solution and the bottom is the red blood cells.
e. Collect the second layer (i.e. lymphocytes) carefully to another tube and wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
f. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
2. Preparation of marrow cells
1) General preparation of marrow cells
a. Aseptic extraction of 0.5 mL bone marrow fluid with heparin anticoagulation.
b. ACK buffer should be restored to room temperature before use.
c. Add 3 mL ACK buffer into the bone marrow fluid and incubate 3~5 min at room temperature.
d. Add 10 mL cell staining buffer (or PBS with 0.1% BSA) to stop lysis.
e. Centrifuge at 300 g for 5 min at room temperature. Discard the supernatant.
f. Repeated the above washing step for another time.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
2) Preparation of mononuclear cells with peripheral blood samples
a. Aseptic extraction of 0.5 mL bone marrow fluid with heparin anticoagulation.
b. Bring the volume to 10 mL with PBS.
c. Add 5 mL (or equal volume of the marrow fluid to be treated) lymphocyte separation solution to the 15 mL conical tube.
d. Add equal volume of peripheral blood along the wall of the tube slowly to make sure that the peripheral blood is above the liquid level of the lymphocyte separation solution. Do not add too fast to prevent the blood from mixing with the separating solution.
e. Centrifuge at 400 g for 20~30 min at room temperature (The centrifuge conditions are according to the blood samples, it is suggested to find out the best separation condition before the experiment). After centrifugation, the blood in the tube should be clearly divided into 4 layers. The upper layer is the plasma, the second is the white Lymphocytes, the third layer is the transparent separation solution and the bottom is the red blood cells.
f. Collect the second layer (i.e. lymphocytes) carefully to another tube and washing the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
3. Preparation of body fluid, lavage fluid or suspension-culture cells
a. Collect the body fluid, lavage fluid or suspension-culture cells in conical tubes.
b. Centrifuge the samples at 300 g for 5 min at room temperature. Discard the supernatant.
c. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature.
d. If there is obvious tissue lumps in suspension, remove the lumps with 200~300 mesh strainers, and wash the cells withcell staining buffer (or PBS with 0.1% BSA) for 1~2 times.
e. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
4. Preparation of adherent cells
a. Remove supernatant of the culture medium and rinse with PBS free of calcium and magnesium ions.
b. Take 25 mL medium as an example, add 1mL digestive solution (0.1% trypsin), digest the cells at room temperature (25°C) or 37°C for2~5 min. Or add 1mL EDTA(0.02%, pH7.4) todigest the cells on ice for 5~10 min.
c. Observe with inverted microscope, if the cytoplasm is retracted and the gap is increased. Removes the digestive solution immediately and add medium with serum to terminate digestion.
d. Absorb the fluid in the bottle with straw and blow the cell wall repeatedly. Please blow gently to avoid bubbles.
e. Transfer to centrifuge tube, centrifuge at 300 g for 5 min at room temperature.
f. Add cell staining buffer (or PBS with 0.1% BSA) to wash the cells 2 times, each time centrifuge at 300 g for 5 min at room temperature, discard supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
5. Preparation of tissue sample
1) Shredding and grinding method
a. Rinse the tissues with PBS or serum-free medium.
b. Place the tissue in a dish and cut it into small particles (1~2 mm3) with ophthalmic scissors.
c. Grind the tissue with syringe plunger to obtain single cell suspension.
d. Mix the cells with appropriate amount of PBS or serum-free medium.
e. Remove tissue lumps with 200~300 mesh strainer bycentrifugation at 300 g for 5 min at room temperature, discard the supernatant.
f. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
2) Mesh screen rubbing
a. Fasten 300 mesh nylon screens on a small beaker.
b. Put the shredded tissue on top of the mesh screen, gently rub the tissue with tweezers and flush with PBS until the tissue is rubbed out.
c. Collect the cell suspension, centrifuge at 300 g for 5 min at room temperature, discard the supernatant.
d. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
e. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
3) Grinding method
a. Cut the tissue into 1~2 mm3 lumps.
b. Put the tissue lumps into grinder, with 1~2 mL PBS added.
c. Grind the tissue until the suspension is homogeneous.
d. Add 10mL PBS to rinse the grinder.
e. Collect cell suspension, and remove tissue lumps by 200~300mesh cell strainer.Centrifuge at 300 g for 5 min at room temperature, discard the supernatant.
f. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
Note:The above methods are mechanical and usually used to treat soft tissues, such as thymus or lymph nodes. For hard tissue or fibrous tissue, the method should be avoided for the cells might be damaged.
For the animal spleen, the red blood cells can be disrupted after grinding. After centrifugation at 300 g for 5 min at room temperature, re-suspend the cell pellet with 3 mL ACK buffer and incubate for 3~5 min at room temperature. Then add 10 mL cell staining buffer (or PBS with 0.1% BSA) to stop lysis. The remaining steps are the same as the grinding method.
4) Trypsin digestion
This method is suitable for tissue with little mesenchyme, such as epithelium, liver and kidney, ect. The calcium ion or serum will inhibit the trypsin digestion, so the buffer containing the ion or serum should be excluded during tissue digestion. The digestion time should be adjusted according to specific conditions. For low temperatures, large tissue messes and low trypsin concentration, the digestion time should be longer. Otherwise, the digestion time will be short. Trypsin is often mixed with EDTA (0.02%) at the rate of 1:1 to improve digestion efficiency. The stepwise protocol is as follows.
a. Rinse the tissue with PBS free of for calcium and magnesium ions.
b. Place the tissue in a dish and cut it into small lumps (1~2 mm3) with ophthalmic scissors.
c. Add trypsin (0.1% trypsin with 0.02% EDTA) to 30 times the volume of the tissue for digestion .
d. Transfer the tissue and digest to a triangular flask with pipette, digest for 20~60 min in 37°Cwater bath or incubator and shake the triangular flask every 5~10 min. If the digestion time is very long, 2/3 of the digestion supernatant can be transferred to a conical tube every 15 min. The cells in supernatants can be protected with ice bath. Or the digestion can be stopped by centrifugation to remove trypsin followed by adding the serum containing medium. New digests are added to the triangle flask to continue the digestion.
e. Filter the digest through 200~300 mesh cell strainer to remove lumps.Centrifuge the digest at 300 g for 5 min at room temperature, discard the supernatant.
f. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
5) Collagen enzyme digestion
This method is suitable for the separation of cells from fibrous tissue, epithelium and cancer tissue. Calcium and magnesium ions can’t inhibit the digestion, so the PBS or serum containing medium can be used to increase cell viability.
a. Rinse the tissue with PBS.
b. Place the tissue in a dish and cut it into small lumps (1~2 mm3) with ophthalmic scissors.
c. Add collagen enzyme containing medium(collagen enzyme at 0.1~0.3 μg/mL) to 30 times the volume of the tissue for digestion.
d. Transfer the tissue and digest to a triangular flask with pipette, digest 4~48 h in 37°Cwater bath or incubator and shake the triangular flask every 5~10 min. If the digestion time is very long, 2/3 of the digestion supernatant can be transferred to a conical tube every 15 min. The cells in supernatants can be protected with ice bath. Or the digestion can be stopped by centrifugation to remove trypsin followed by adding the serum containing medium. New digests are added to the triangle flask to continue the digestion.
e. Filter the digest through 200~300 mesh cell strainer to remove lumps.Centrifuge the digest at 300 g for 5 min at room temperature, discard the supernatant.
f. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
g. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
Note:These are the most common sample preparation methods for the tissues. Mechanical methods may cause a certain degree of cell damage and low single cell production. While enzyme and chemical methods (enzyme mixed with EDTA) may have unpredictable effects on the chemical composition of the cells though they are better for dispersing and depolymerizing the solid tissue. Choose a suitable method for the single cell suspension preparation according to the experimental purposes.
6. Preparation of paraffin-embedded tissue samples for flow cytometry
Most tissues obtained from surgery are paraffin-embedded. The preparation of single cell suspension from paraffin-embedded tissue expands the scope of application for flow cytometry.
a. Cut 3~5 tissue slices of 40~50 μm thick off from the paraffin-embedded tissue. Or use the mortar to grind the paraffin-embedded tissue to lumps of 0.5 mm in diameter. Put the slices or the lumps into a 10 mL tube.
b. Add 5~8 mL xylene to the tube, for dewaxing at room temperature for 1~2 days. Replace the xylene for 1~2 times until the paraffin wax is moved out completely. Discard the xylene.
c. Hydration: Replace the medium in order with 5 mL 100%, 95%, 70%, 50% ethanol gradient, 10 min each time.
d. Replace the ethanol with 3~5 mL distilled water 3~5 mL for 10 min, then discard the water.
e. Digestion: Add 2 mL 0.5% trypsin digestion trypsin (pH1.5~2.0) to digest the tissue for 30 min at 37°C. During this period, shake the tube every 10 min.
f. Add the serum containing medium to terminate digestion.
g. Remove the undigested lumps with 300 mesh strainer.Undigested tissue can be digested for the second time.
h. Centrifuge the digest at 300 g for 5 min at room temperature, discard the supernatant.
i. Wash the cells with cell staining buffer (or PBS with 0.1% BSA) for 2 times, each time by centrifugation at 300 g for 5 min at room temperature, discard the supernatant.
j. Re-suspend the cells with cell staining buffer (or PBS with 0.1% BSA) and adjust the concentration to 1X107/mL.
Notice
a. Fresh tissue specimens should be treated and preserved in time to avoid the tissue necrosis or cell self-melting after placed at room temperature for long to time which will affect the results of FCM.
b. When enzymatic method is used, please pay attention to the conditions and influence factors. The influence of enzyme solvent, digestion time, pH value and concentration of the enzyme will affect the results.
c. Specific methods should be chosen for different tissues to get high quality mono-dispersed cells. For example, if the tissues are rich in cells such as lymphosarcoma, optic neuroblastoma, brain tumor, undifferentiated tumor, medullary tumor, and some soft tissue sarcoma, the simple mechanical methods will be a recommended choice to obtain massive high-quality mono-dispersed cells.
d. For enzymological methods, we should pay attention to the enzyme used. For example, tumors rich in connective tissues, such as esophageal, breast and skin cancers, collagen enzyme should be the best choice.