|
Concentration of separating gel |
Linear separation range |
| 6% | 50-150 kDa |
| 8% | 30-90 kDa |
|
10% |
20-80 kDa |
| 12% | 12-60 kDa |
| 15% | 10-40 kDa |
Western Blot
WB
| Components | Sample volume of each component corresponding to different stacking gel volumes | |||||||
| 1 mL | 2 mL | 3 mL | 4 mL | 5 mL | 6 mL | 8 mL | 10 mL | |
| 5% stacking gel | ||||||||
| H2O | 0.68 | 1.4 | 2.1 | 2.7 | 3.4 | 4.1 | 5.5 | 6.8 |
| 30% Acrylamide | 0.17 | 0.33 | 0.5 | 0.67 | 0.83 | 1.0 | 1.3 | 1.7 |
| 1.0M Tris-HCl(pH 6.8) | 0.13 | 0.25 | 0.38 | 0.5 | 0.63 | 0.75 | 1.0 | 1.25 |
| 10% SDS | 0.01 | 0.02 | 0.03 | 0.04 | 0.05 | 0.06 | 0.08 | 0.1 |
| 10% (NH4)2S2O8 | 0.01 | 0.02 | 0.03 | 0.04 | 0.05 | 0.06 | 0.08 | 0.1 |
| TEMED | 0.001 | 0.002 | 0.003 | 0.004 | 0.005 | 0.006 | 0.008 | 0.01 |
| Components | Sample volume of each component corresponding to different separation gel volumes | |||||||
| 5 mL | 10 mL | 15 mL | 20 mL | 25 mL | 30 mL | 40 mL | 50 mL | |
| 6% Gel | ||||||||
| H2O | 2.6 | 5.3 | 7.9 | 10.6 | 13.2 | 15.9 | 21.2 | 26.5 |
| 30% Acrylamide | 1.0 | 2.0 | 3.0 | 4.0 | 5.0 | 6.0 | 8.0 | 10.0 |
| 1.5M Tris-HCl(pH8.8) | 1.3 | 2.5 | 3.8 | 5.0 | 6.3 | 7.5 | 10.0 | 12.5 |
| 10% SDS | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| 10% (NH4)2S2O8 | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| TEMED | 0.004 | 0.008 | 0.012 | 0.016 | 0.02 | 0.024 | 0.032 | 0.04 |
| 8% Gel | ||||||||
| H2O | 2.3 | 4.6 | 6.9 | 9.3 | 11.5 | 13.9 | 18.5 | 23.2 |
| 30% Acrylamide | 1.3 | 2.5 | 4.0 | 5.3 | 6.7 | 8.0 | 10.7 | 13.3 |
| 1.5M Tris-HCl(pH8.8) | 1.3 | 2.5 | 3.8 | 5.0 | 6.3 | 7.5 | 10.0 | 12.5 |
| 10% SDS | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| 10% (NH4)2S2O8 | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| TEMED | 0.003 | 0.006 | 0.009 | 0.012 | 0.015 | 0.018 | 0.024 | 0.03 |
| 10% Gel | ||||||||
| H2O | 1.9 | 4.0 | 5.9 | 7.9 | 9.9 | 11.9 | 15.9 | 19.8 |
| 30% Acrylamide | 1.7 | 3.3 | 5.0 | 6.7 | 8.3 | 10.0 | 13.3 | 16.7 |
| 1.5M Tris-HCl(pH8.8) | 1.3 | 2.5 | 3.8 | 5.0 | 6.3 | 7.5 | 10.0 | 12.5 |
| 10% SDS | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| 10% (NH4)2S2O8 | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| TEMED | 0.002 | 0.004 | 0.006 | 0.008 | 0.01 | 0.012 | 0.016 | 0.02 |
| 12% Gel | ||||||||
| H2O | 1.6 | 3.3 | 4.9 | 6.6 | 8.2 | 9.9 | 15.9 | 16.5 |
| 30% Acrylamide | 2.0 | 4.0 | 6.0 | 8.0 | 10.0 | 12.0 | 13.3 | 20.0 |
| 1.5M Tris-HCl(pH8.8) | 1.3 | 2.5 | 3.8 | 5.0 | 6.3 | 7.5 | 10.0 | 12.5 |
| 10% SDS | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| 10% (NH4)2S2O8 | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| TEMED | 0.002 | 0.004 | 0.006 | 0.008 | 0.01 | 0.012 | 0.016 | 0.02 |
| 15% Gel | ||||||||
| H2O | 1.1 | 2.3 | 3.4 | 4.6 | 5.7 | 6.9 | 9.2 | 11.5 |
| 30% Acrylamide | 2.5 | 5.0 | 7.5 | 10.0 | 12.5 | 15.0 | 20.0 | 25.0 |
| 1.5M Tris-HCl(pH8.8) | 1.3 | 2.5 | 3.8 | 5.0 | 6.3 | 7.5 | 10.0 | 12.5 |
| 10% SDS | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
| 10% (NH4)2S2O8 | 0.05 | 0.1 | 0.15 | 0.2 | 0.25 | 0.3 | 0.4 | 0.5 |
Western Blot result depends on the whole system including antigen content, sensitivity of primary antibody, sensitivity of secondary antibody, sensitivity of substrate, efficiency of color development and photographic fixing. Mistake of any procedure in the experiment may lead to unsatisfactory results.
Here Elabscience lists the common Western Blot troubleshooting.
- No band or low band
| Possible causes | Suggestions |
| The loading amount of sample is low | Increase the loading amount |
| The target protein quantity is too low or not expressed in the sample | Refer to the relevant literatures to make sure that the sample contain your target protein, or prepare fresh sample again and choose a positive sample as control |
| There is a poor transfer of protein to membrane or not enough protein is bound to the membrane | Ensure that the transfer order is correct and the transfer time is sufficient |
| The antibody concentration may be too low | Use a higher concentration of antibody |
| The antibody concentration may be too high to cause the signal to disappear instantly | Use a lower concentration of antibody |
| Insufficient exposure time | Prolong the exposure time |
| Insufficient incubation or deactivation of the substrate | Increase the incubation time of substrate and ensure that the substrate is valid |
| The target protein transferred to the membrane has degraded | Keep a low transfer temperature, decrease the transfer electric current and transfer time |
| Excessive washing of the membrane | Reduce the frequency or duration of washing steps |
| The antibody is inactive or the titer of antibody is too low | Pay attention to the preservation of antibody and use antibody with higher titer |
- High background
| Possible causes | Suggestions |
| The experimental equipment has been contaminated | Ensure that the equipment is clean |
| Some membrane may cause high background | NC membranes are considered to cause less background than PVDF membranes. |
| Blocking buffer is not compatible or there is cross-reactivity between the blocking buffer with antibodies | Replace the blocking buffer |
| Insufficient blocking | Prolong the blocking time |
| The antibody concentration may be too high | Use a lower concentration of antibody |
| Insufficient washing | Increase the frequency and duration of washing |
| Excessive exposure time | Shorten the exposure time |
| The membrane or buffer has been contaminated | Use the fresh buffer and keep the membrane moist during the experiment |
- Non-specific bands
| Possible causes | Suggestions |
| The protein sample has digested during the treatment | Choose fresh samples for experiment |
| The loading amount of sample is too much | Reduce the loading amount |
| Insufficient blocking | Prolong the blocking time |
| The washing of membrane may be insufficient | Ensure sufficient washing of membrane |
| The antibody concentration may be too high | Use a lower concentration of antibody |
| The antibody specificity is low. | Use antibody with good specificity |
| The target protein has multiple spliceosomes or modified sites | Refer to relevant literatures to check whether the target protein has other spliceosomes or modified sites |
- Other problems
| Problems | Possible causes | Suggestions |
| Black dots on the membrane | The antibodies may have non-specifically binding with the blocking buffer | Replace the blocking buffer |
| White bands | The target protein content is too high or antibody concentration is too high | Reduce the loading amount or decrease the concentration of the antibody |
| Molecular weight is very low or high | Inappropriate gel percentage or uneven gel. /The electrophoresis temperature may be too high | Change the gel percentage: use a higher percentage for small proteins and a lower percentage for large proteins |
| Uneven bands/ Bands trail or deviate or diffuse to both sides | The equipment is not suitable. / There is bubble at the bottom/ The sample is not dissolved well. / The electrode is not balanced. / The sample amount is too much | Ensure that the electrophoresis gel is in good state and horizontal position/ Ensure the sample extraction/ Reduce the sample amount |
Summary: The Western blot technology is rather mature, but it is not easy to get the desired result with just one trial. It is recommended to explore and optimize the experimental conditions, thus to get your ideal Western bands in the formal experiment.
Sample preparation
1. Protein extraction
1) Experimental instruments and reagents:
Homogenizer, RIPA buffer, PMSF (proteinase inhibitor), phosphatase inhibitor, scissors, tweezers, alcohol cotton, PBS, pipette, centrifugal machine.
RIPA preparation—Add 1μL PMSF and phosphatase inhibitor in each 100μL RIPA. Prepare when needed. Calculated the total volume of RIPA buffer before treating the samples.
2) For tissue sample:Sterilize scissors and tweezers with alcohol cotton, wash the tissue with PBS, add appropriate amount of tissue into homogenizer, cut the tissue into pieces and add enoughRIPA buffer, then homogenize in ice water bath for 1-4 minutes. After homogenizing, transfer the solution to an EP tube and centrifuge at 12000rpm for 5 minutes, take supernatant.
3) For cell sample:
A) Adherent cells:Take out and wash the culture medium with PBS. Add appropriate amount of RIPA buffer (150μL RIPA buffer per 5X10e6 cells), Crack on ice for 5-10 minutes, pipette several times until cell desquamate. Then pipette cracking solution into EP tube. Centrifuge at 12000rpm for 5 minutes, take supernatant. For adherent cells in large bottle, use pancreatin to elute them and RIPA buffer afterwards. The lysis method refers to suspension cell processing.
B)Suspension cell:Add cells in an EP tube and centrifuge at 2000-3000 rpm for 5 minutes, remove the supernatant medium. Add 100-150μLRIPA bufferto each 20μLcells, pipette several times until cells disrupt completely.Centrifuge at 12000rpm for 5 minutes, take supernatant.
The protein can be stored for one week in 4℃, for 2 months in -20℃, and for 6 months in -80℃.
2. Measurement of protein concentration
1) Experimental instruments and reagents:
ELISA microplate, Microplate reader, centrifuge, 1 mg/mL BSA, BCA reagent, PBS.
2) Standard curve:use total volume 20μL of BSA and PBS to make the standard curve, operate according to the follows.
Dilute BSA protein standard into serial concentrations: 0, 0.2, 0.4, 0.6, 0.8, 1.0 mg/mL (0, 4μL, 8μL, 12μL, 16μL, 20μL BSA, add up to 20μL with PBS), set duplicates for each gradient.
3) Dilute sample to test the concentration
Ratio of 10 times: take 18μL PBS and 2μL sample supernatant, set triplicates for each sample.
Ratio of 20 times: take 19μL PBS and 1μL sample supernatant, set triplicates for each sample.
The total volume is 20μL.
4) BCA reagent preparation:Prepare fresh solution before use, avoid of being contaminated by protein. (Detailed steps reference instructions)
5) Incubation:Add 200μL BCA to standard and sample wells, incubate at 37℃for 15-20 minutes.
Read the OD value at 568nm with microplate reader. Average the standard OD value, minus the blank OD value, then make the standard curve and standard curve equation. Average the sample OD value, calculate according to the equation to get the protein concentration.Use the calculation to get the loading volume of samples. Recommend total protein amount of each sample is 50ug.
Note:All OD value must minus the blank OD value before calculating.
3. Sample denaturation
Mix sample supernatant and 5× SDS buffer according to the ratio 4:1, incubate in boiling water bath for 10 minutes. Samples with high concentration can be diluted with PBS before boiling, loading volume of samples should be near 10ul.
Store the processed sample at -20℃(validity for 6 months), -80℃for long time.
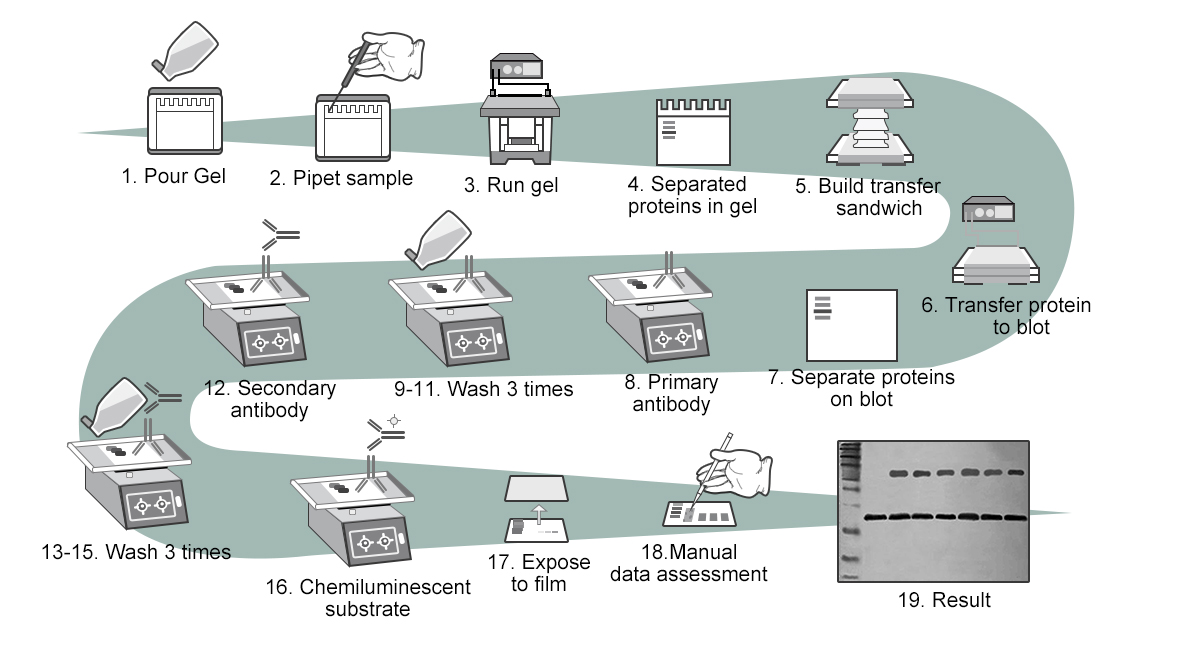
WB Experiment Procedure
1. Gel preparation
| Molecular weight of protein(kDa) | Concentration of separation gel(%) |
| >140 | 6% |
| 100-140 | 8% |
| 30-100 | 10% |
| 10-30 | 12% |
| <10 | 15% |
1) Prepare separation gel according to the molecular weight of protein. Shake slowly to mix completely, avoid oxygen in when shaking hard. Pour the mixed gel in between two glass panes, inject anhydrous ethyl alcohol above it to avoid oxygen in to affect polymerization.
2) Remain horizontal, incubate at 37℃for 1 hour till it polymerizes completely.
3) Prepare thestackinggel according to proportion. Shake slowly to mix completely, pour out the anhydrous ethyl alcohol above separation gel, and absorb the anhydrous ethyl alcohol above solidified separation gel with filter paper. Pour spacer gel gently, insert comb carefully and avoid bubbles.
4) Incubate at 37℃for 1 hour till it polymerizes completely, then take out the comb.
2. Adding samples
Add electrophoretic buffer to electrophoresis tank, the buffer solution must be above sample wells, the bottom of gel must be immersed by buffer solution. Level within glass panes should be above level outside. Add samples according to the calculated loading volume and add pre-stained protein marker, make sure the total protein in every well is between 30-50μg, the total loading volume of samples should be less than 30μL.
Make sure that samples are added in short time, and avoid of samples splash.
3. Electrophoresis
Cover the lid of electrophoresis tank, mind the positive and negative, and choose the appropriate voltage to do electrophoresis. Usually in inconsecutive system, the voltage ofstackinggel (80V is suggested) is lower than the voltage of separation gel (110-150V is suggested). Sample will be at the same level when into the separation gel. Electrophoresis time is about 2-3 hours till bromophenol blue reaches the bottom of gel.
4. Wet transfer
1) Take out the gel after electrophoresis, rinse in the cold electrophoretic buffer solution for few seconds. According to the sandwich mode, open the Electrical transfer folder, stack up a sponge mat soaked by transferring buffer at both sides. Then place the qualitative filter paper soaked by transferring buffer at both sides. Place the gel flat on the negative filter paper, and then place flat the PVDF membrane (the PVDF is soaked in methyl alcohol for 5minutes, then saturated in transferring buffer) on the gel. Wipe off the bubbles, and fold the folder. Note that wipe off all the bubbles.
2) Load the transferring buffer in transfer tank, insert the folder. Put the tank in ice water, make sure PVDF membrane is near the positive pole, amino acid and protein with electronegative will move to the positive pole.
3) Choose the constant current, the electric current of each tank is suggested to be 150-200mA. Adjust the time according to molecular weight of the protein.
4) After transferring, take out the transferred PVDF membrane, rinse the membrane with washing buffer at room temperature.
5. Blocking
Take out the rinsed membrane, put it into the blocking buffer (5% skim milk powder or 5% BSA). Incubate on the shaker atroom temperaturefor 2 hours.
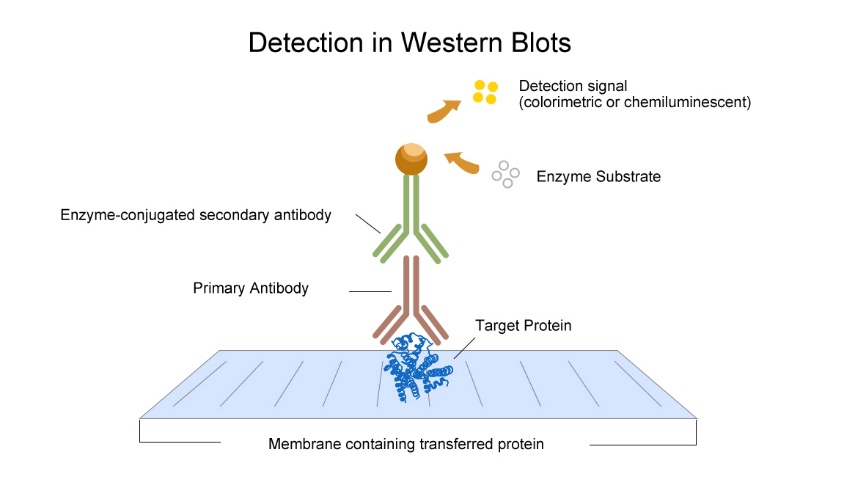
6. Incubation of antibody
The indirect method is recommended. Add unlabeled primary antibody, the antibody binds to the antigen protein. Then add the enzyme labeled secondary antibody to test.
1) Add diluted primary antibody to blocked membrane, incubate over night at 4℃.
2) Wash the membrane with washing bufferfor 10 minutes and repeat 3 times.
3) After adding the diluted secondary antibody, incubate at room temperature for 2 hours. Wash the membrane for 10 minutesand repeat 3 times.
7. Detection
The detection method depends on the label at secondary antibody. ECL and DAB are usually used.
1)ECL method
Use HRP to catalyze chemiluminescent compound, an intermediate is formed, record the luminescence withmachine. Also we can use X-ray film to fixate the images and use DAB as colour developing reagent.
A) ECL preparation: mix the substrate A and B as the ratio 1:1. Prepare when needed.
B) Cover the blotting membrane with mixed substrate for 1-5 minutes, observe the fluorescence in dark.
C) Record the result with machine or in dark room with autoradiography film or chemiluminiscence imaging system.
D) For autoradiography film, expose for several seconds to minutes according to the fluorescence intensity. The exposed film will be soaked in developing liquid till strips appear, then rinse in the fixing solution. Wash the film with water and hang up to dry.
2)DAB Detection
A) Add 1mL water and a drop ofchromogenicreagent A, B, C, mix fully.
B) Chromogenic reaction: put appropriate amount of DAB flat on blotting membrane, observe in room temperature, obvious brown protein strip will appear.
8. Data analysis
Analyze the bands with Bandscan software
1.0 mol/L Tris•HCl(pH6.8)
| Tris | 30.28 g |
| Ultrapure water | 200 mL |
Dissolve the Tris in the ultrapure water, and then add concentrated HCl to adjust the pH to be 6.8. Add ultrapure water until the total volume is 250 mL. Store the prepared buffer at room temperature after high temperature sterilization.
1.5 mol/L Tris•HCl(pH8.8)
| Tris | 45.43 g |
| Ultrapure water | 200 mL |
Dissolve the Tris in the ultrapure water, and then add concentrated HCl to adjust the pH to be 8.8. Add ultrapure water until the total volume is 250mL. Store the prepared buffer at room temperature after high temperature sterilization.
10% SDS
Dissolve 10g SDS in 100 mL of distilled water by incubating in 50℃ water bath, and store the prepared buffer at room temperature. If there is any precipitate during the long time storage, the buffer can still be used after dissolution by water bath.
10% Ammonium persulfate(APS)
Dissolve 0.1g ammonium persulfate in 1.0 mL of ultrapure water, and then store the prepared buffer at 4℃ for 1 week (-20℃ for long time storage).
30% Acrylamide
| Acrylamide | 29 g |
| N,N’-Methylenebisacrylamide | 1 g |
Dissolve the acrylamide and N,N’-Methylenebisacrylamide in 50 mL water. Then add distilled water until the total volume is 100 mL. Store the prepared buffer at room temperature in brown bottle.
10× PBS (2L)
| NaCl | 160 g |
| KCl | 4 g |
| Na2HPO4·12H2O | 22.9 g |
| KH2PO4 | 4 g |
| Distilled water | 1 L |
Mix all of the substances above homogeneously and then add NaOH to adjust the pH to be 7.4. Add distilled water until the total volume is 2L. Store the buffer at room temperature.
Reduced 5×SDS (Loading Buffer)
| 0.5 mol/L Tris•HCl (pH6.8) | 2.5 mL |
| Dithiothreitol (DTT) | 0.39 g |
| SDS | 0.5 g |
| Bromophenol Blue | 0.025 g |
| Glycerin | 2.5 mL |
Mix all of the substances above homogeneously, and then aliquot the buffer and respectively. Store them in 1.5 mL centrifugal tubes at 4℃ for weeks(-20℃ for long time storage).
Electrophoretic Buffer
| Tris | 3.03g |
| Glycine | 18.77g |
| SDS | 1g |
Add distilled water to dissolve the substances above until the total volume is 1000 mL. Store the buffer at room temperature. The prepared solution can be repeatedly used for 3 to 5 times.
Transfer Buffer
1×Transfer Buffer:
| Glycine | 11.25 g |
| Tris | 2.375 g |
| SDS | 0.375 g |
| Methanol | 200 mL |
Add distilled water to dissolve the substances above until the total volume is 1000 mL. Store the buffer at 4℃. The prepared solution can be repeatedly used for 3 to 5 times.
TBS Buffer
| NaCl | 8.5 g |
| Tris | 1.2 g |
| SDS | 0.375 g |
| Methanol | 200 mL |
Add distilled water to dissolve the NaCl and Tris, SDS, and then add glacial acetic acid to adjust the pH to be 7.2. Add 200mL Methanol to the buffer then add distilled water until the total volume is 1000mL. Store the buffer at room temperature.
Eluant TBST Solution
Mix 1000 mL of TBS solution with 1 mL of Tween-20, then homogeneously mix the buffer. Prepare the buffer before when needed.
Blocking Buffer(TBST buffer with 5% skimmed milk)
| Skimmed milk | 5 g |
| TBST | 100 mL |
Dissolve the skimmed milk in TBST. The prepared TBST buffer can be used for only one time.
RIPA Lysis Buffer, Medium
| Reagent | Amount | Final concentration |
| Tris-base | 0.6055 g | 50 mM |
| NaCl | 0.8766 g | 150 mM |
| EDTA·2Na.2H2O | 0.037224 g | 1 mM |
| SDS | 0.1 g | 0.1% |
| Sodium deoxycholate | 0.5 g | 0.5% |
| NP-40 | 1 mL | 1% |
Dissolve Tris-base, NaCl, EDTA and SDS with distilled water, and then add concentrated HCI to adjust the pH to be 7.4. Add 5 mL of the sodium deoxycholate solution of 10% that is prepared separately in advance (the working concentration of sodium deoxycholate solution is 0.5%), and add 1 mL of NP-40. Mix them and add distilled water until the total volume is 100 mL. Aliquot the buffer and store at -20℃.
Western Blotting is used to identify the large molecular antigen (usually protein) that can interact with specific antibodies and determine the MV of the antigen. The protein was separated by SDS electrophoresis gel, and then transferred to solid phase support by electrophoresis. Solid phase support includes nitrocellulose membrane, poly vinylidene fluoride two (PVDF) membrane and cationic nylon membrane. The unreacted sites on the membrane were sealed up to inhibit the nonspecific adsorption, so that the immobilized proteins could interact with specific primary antibodies and then the secondary antibodies which conjugated with enzyme or fluorescein. Finally, it is located by means of radiation, chromophore or chemiluminescence.
Western Blot reagents
- 1.0 mol/L Tris•HCl(pH6.8)
- 1.5 mol/L Tris•HCl(pH8.8)
- 10% SDS
- 10% Ammonium persulfate(APS)
- 30% Acrylamide
6 Reduced 5×SDS (Loading Buffer)
- Electrophoretic Buffer
- Transfer Buffer
- TBS Buffer
- 10.Eluant TBST Solution
- Blocking Buffer(TBST buffer with 5% skimmed milk or 5% BSA)
- Antibody dilution buffer: Usually using blocking buffer as antibody dilution buffer which contain 5% skimmed milk or 5% BSA, Each antibody has its best dilution ratio.
Steps of the experiment
1.Prepare the gel(separation gel) according to the molecular weight of protein.
- Using appropriatelysates and methods to prepare samples. Adding samples.
- Electrophoresis:Usually in inconsecutive system, the voltage ofstacking gel (80V is suggested) is lower than the voltage of separation gel (110-150V is suggested)
- After Electrophoresis, cut off the useless gel which has no sample, thesponge mat and filter paper soaked by transferring buffer.
- Transfer the proteins to membrane:
Choose appropriate membrane: take PVDF membrane as example
Prepare PVDF membrane, soak in Methanol for 5s
Put black plate – fiber mats – filter paper – gel -PVDF membrane – filter paper – fiber mat – white plate in order successively.
6.Choose the constant current, the electric current of each tank is suggested to be 150-200mA. Adjust the time according to molecular weight of the protein.
- Blocking:Take out the rinsed membrane, put it into the blocking buffer (5% skim milk powder or 5% BSA). Incubate on the shaker at room temperature for 2 hours.
- Incubation of antibody: Add diluted primary antibody to blocked membrane, incubate over night at 4℃.
- 9.Wash the membrane with washing bufferfor 10 minutes and repeat 3 times.
- Add the diluted secondary antibody, incubate at room temperature for 2 hours.
- Wash the membrane for 10 minutesand repeat 3 times.
- ECL preparation: mix the substrate A and B as the ratio 1:1. Prepare when needed.
- Cover the blotting membrane with mixed substrate for 1-5 minutes, observe the fluorescence in dark.
- Record the result with machine or in dark room with autoradiography film or chemiluminiscence imaging system.
- For autoradiography film, expose for several seconds to minutes according to the fluorescence intensity. The exposed film will be soaked in developing liquid till strips appear, then rinse in the fixing solution. Wash the film with water and hang up to dry.
- Analyze the bands with Bandscan software.
Your first choice for scientific solutions
Find out moreSUPPORT
outstanding technical support
PRODUCT
we offer a full product guarantee
DELIVERY
we offer free delivery to UK universities and non profit organisations