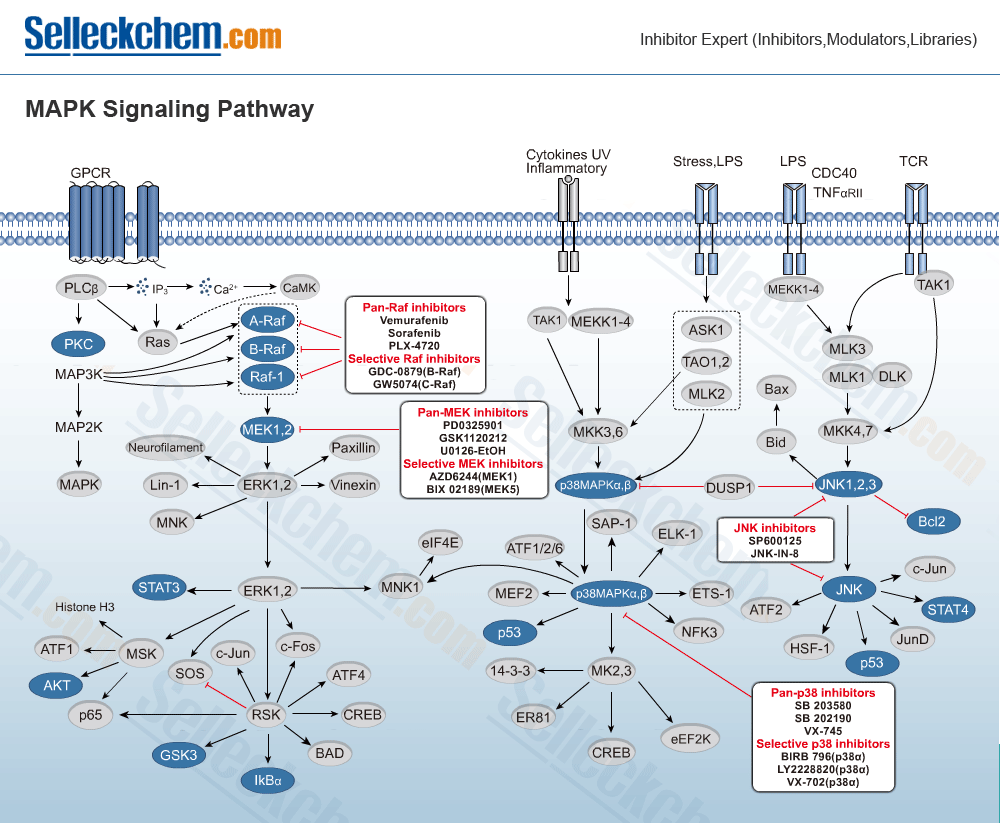
MAPK (mitogen-activated protein kinase) is a family of serine-threonine kinases that regulate multiple cellular functions such as mitogens, proinflammatory cytokines, gene expression and cell survival/apoptosis. ERK1/2, p38, ERK3/4, ERK5, ERK7/8, JNKs, and SAPKs have all been identified in the MAPK family.
Selleck Chemicals supply products to the following targets:
- MEK
- Raf
- P38 MAPK
- JNK
- ERK
The mitogen-activated protein kinase (MAPK) is the backbone of four major signal transduction cascades leading to the phosphorylation and activation of extracellular signal-regulated kinases 1 and 2 (ERK1-2), JNK, p38 and ERK5. MAPK 1 and 2 are typically referred to as MEK1 and 2 and share ~80% structural homology. These dual-specificity tyrosine/threonine protein kinases are active in ~30% of all human cancers where the MAPK signalling pathway is implicated.[1] MEK exists just downstream of RAF in the classical MAPK pathway known as RAS-RAF-MEK-ERK pathway. Phosphorylation of MEK by RAF results in the phosphorylation of ERK1 and ERK2 – these appear to be the only targets of MEK. The phosphorylation of ERK modifies the activity of a number of transcription factors resulting in altered gene expression and therefore cell proliferation, typically blocking apoptosis. It is involved in a number of other processes that drive cancer progression, such as cell motility, metastasis, and angiogenesis.[2]
The activation of the MAPK signalling pathway begins when growth factors, hormones and chemokines interact with respective tyrosine kinase receptors. Frequently, it is observed that the MAPK pathway is activated in tumors where receptor tyrosine kinases, such as EGFR, are over expressed due to somatic mutation, gene amplification and/or increased autocrine or paracrine signalling. Upon ligand binding to the extracellular domain of the receptor, the receptor tyrosine kinases dimerise and induce activation of kinase activity in the cytoplasmic domain, leading to phosphorylation of C-terminal tyrosine residues. This provides docking sites for proteins containing Src homology 2 (SH2) or phosphotyrosine binding domains, such as adaptor protein growth factor receptor-bound protein 2 (GRB2). Following these events, the adaptor proteins recruit further effectors, including the guanine-nucleotide exchange factor, SOS (also known as son of sevenless). In the plasma membrane, SOS is recruited and nucleotide exchange of GDP for GTP occurs with RAS proteins (KRAS, NRAS, and HRAS). As a consequence, GTP-bound RAS activates the closely related RAF protein kinases (CRAF, BRAF, and ARAF). Recruitment of RAF to the plasma membrane facilitates the completion of the RAF catalytic kinase activity including the phosphorylation of the MAPK kinases (MAPKK), MEK1 and MEK2, dual-specificity kinases that recognise and phosphorylate tyrosine and threonine residues in the Thr-X-Tyr activation loop of the MAPKs. Activated ERK1 and ERK2 phosphorylate cytosolic signalling proteins such as p90 ribosomal S6 kinase (RSK) and MAPK-interacting serine/threonine kinase (MNK), and transcription factors (Elk-1, CREB, c-Fos and c-Jun).[3]
Aside from constitutive receptor tyrosine kinase over expression, constitutive activation of the RAS-RAF-MEK-ERK pathway can result from mutations in RAS, BRAF, and/or MEK genes. Mutations in RAS family genes (HRAS, KRAS, and NRAS) are observed to be a result of single-amino-acid substitutions in codons 12, 13, and 61. BRAF mutations are common in the P-loop (exon 11) and in the activation segment (exon 15) of the kinase domain. The substitution of a valine residue at position 600 for glutamic acid (V600E) is found in an overwhelming majority of BRAF mutations involved in human cancers. The majority of BRAF mutations destabilise the inactive conformation of the protein, disrupting the interaction between the P-loop and the activation segment.[2]
Constitutive activation of the kinase signalling pathway, RAS-RAF-MEK-ERK, has been associated with cancers of the lung, colon, melanoma, lung, thyroid, leukemia, and pancreatic cancer. As a consequence there is great interest in targeting the RAS-RAF-MEK-ERK axis in oncology drug development. In addition, there appears to be crosstalk between RAS-RAF-MEK-ERK with other signalling pathways. For example, RAS can activate the PI3K-AKT pathway, aside from having shared inputs, and there appears to be some compensation for signalling activity when one or the other is inhibited. Research has shown that when mTOR is inhibited, MAPK activity can still occur as a result of PI3K activation through RAS. Dual activation of these two pathways is observed in a number of cancer types including melanoma, prostate and colorectal cancer, and provides the rationale for combining therapeutic agents.[2]
MAPK inhibitors include compounds targeting MEK – the first MEK inhibitor, CI-1040 (PD-184352), was tested in humans in 2000 through a joint partnership between Pfizer and Warner-Lambert. However, the compound was not shown to be effective following a Phase II clinical study that tested the compound in a range of cancers (breast cancer, small cell lung cancer (SCLC), pancreatic cancer, and colon cancer). Based on the concept of CI-1040, Pfizer explored the development of a similar compound called PD-0325901 that was touted to have improved biological and pharmaceutical properties than its precursor. However, the compound failed in a Phase II clinical trials for evaluation against non-small cell lung cancer (NSCLC), breast cancer, colon cancer, and melanoma, due to safety concerns. Aside from Pfizer, other pharmaceutical companies have developed compounds targeting the MAPK signalling pathway including AstraZeneca and Array BioPharma. Together, these two companies led the development of ARRY-142886/AZD6244 as a monotherapy for melanoma, colon cancer, NSCLC, and pancreatic cancer. Bayer has also explored targeting MEK as an anti-cancer strategy with the development of BAY869766/RDEA119 in combination with Sorafenib for the treatment of liver cancer. In addition, the pairing of BAY869766 with Gemcitabine for the treatment of pancreatic cancer has also been investigated by Bayer. Collaboration between Genentech and Exelixis is ongoing for the evaluation of GDC0973/XL518 in combination with GDC0941 for advanced/metastatic tumors. Meanwhile, Hoffmann-La Roche has ventured into the targeting of MEK with the development of RO4987655 and R05126766. However, it should be noted that neither compound has advanced to Phase II. And lastly, GlaxoSmithKline, EMD Serono, and Takeda have also explored the development of MEK inhibitors (GSK1120212, AS703026, and TAK-733 respectively). In all the clinical activity to date, the most popular approach to the utility of MEK inhibition appears to be exploring MEK inhibitors in combination with other kinase inhibitors in the hopes of discovering synergistic effects with other compounds. The choice to explore a co-inhibitory strategy is likely due to the cytostatic effects of MEK inhibitors; in contrast other anti-tumor compounds have more cytotoxic effects on cancer cells. Despite the lack of clinical advancement of MEK inhibitors leading to an FDA-approved compound, MEK inhibitors continue to show promise not only as an anti-tumor strategy by targeting MAPK activity, but also for their potential use in immune disorders.[4]
References
- Inamdar, GS, Madhunapantula, SV, and Robertson, GP. Targeting the MAPK pathway in melanoma: Why some approaches succeed and others fail. Biochem. Pharm. 2010;80: 624-637.
- Chapman, MS, and Miner, JN. Novel mitogen-activated protein kinase kinase inhibitors. Expert Opin. Investig. Drugs 2011;20(2): 209-220.
- De Luca, A, Maiello, MR, D’Alessio, A, Pergameno, M, and Normanno, N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin. Ther. Targets 2012;16S: S17-S27.
- Trjuillo, JI. MEK inhibitors: a patent review 2008 – 2010. Expert Opin. Ther. Patents 2011;21(7): 1045-1069.